Gitelmanův syndrom: jak ho poznat a jak jej odlišit od jiných příčin hypokalemie?
Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) consensus conference. Kidney International 2017;91:24–33.
Gitelmanův syndrom (GS) je relativně vzácné autozomálně recesivně dědičné onemocnění, které patří mezi tzv. salt‑losing nephropathies, tedy tubulopatie s vysokými ztrátami soli do moči. S prevalencí výskytu 1–10/40 000 jedinců patří mezi nejčastější vrozená tubulární onemocnění (častější je výskyt v Asii). Typicky se GS projevuje jako hypokalemická metabolická alkalóza spolu s hypomagnezemií a hypokalciurií. Hypomagnezemie však může u některých jedinců chybět a výskyt hypokalciurie bývá variabilní (od velmi nízkých až skoro k normálním hodnotám exkrece kalcia). Onemocnění je způsobeno inaktivačními mutacemi v genu SLC12A3, a pokud postihuje obě alely, dochází k poruše funkce thiazid‑senzitivního Na+‑Cl– kotransportéru (NCCT) lokalizovaného v apikální membráně tubulárních buněk distálního stočeného kanálku. Do současné doby bylo identifikováno kolem 350 různých mutací v tomto genu. Většina nemocných jsou heterozygoti, ale u celé řady nemocných s jasnou klinickou manifestací byla nalezena mutace jen na jedné alele.
Gitelmanův syndrom je považován za benigní formu „salt‑losing“ nefropatií, které se většinou začnou manifestovat v adolescentním až dospělém věku. Zvýšené ztráty Na+ a Cl– způsobují hypovolemii a sekundární aktivaci renin‑angiotenzin‑aldosteronového systému (RAAS). V důsledku toho se kompenzatorně zvyšuje reabsorpce Na+ ve sběrných kanálcích vybalancovaná zvýšenou sekrecí iontů K+ a H+, což je příčinou hypokalemické alkalózy. I přes relativně benigní průběh onemocnění s malými či žádnými symptomy choroby někteří autoři poukazují na to, že GS může vést ke zhoršení kvality života nemocných a také ke zvýšenému výskytu depresí u těchto pacientů.1
Zvýšená exkrece Mg2+ do moči je způsobena snížením jeho reabsorpce v distálním stočeném kanálku kanálem TRPM6, jehož exprese je u pacientů s GS defektní (etiologie není zcela jasná, ale je zde možný spolupodíl mutací v genu pro claudin‑16).2 Vzhledem k tomu, že se tento kanál nachází i v duodenu, ovlivňuje hypomagnezemii pravděpodobně i snížená resorpce Mg2+ ve střevě.
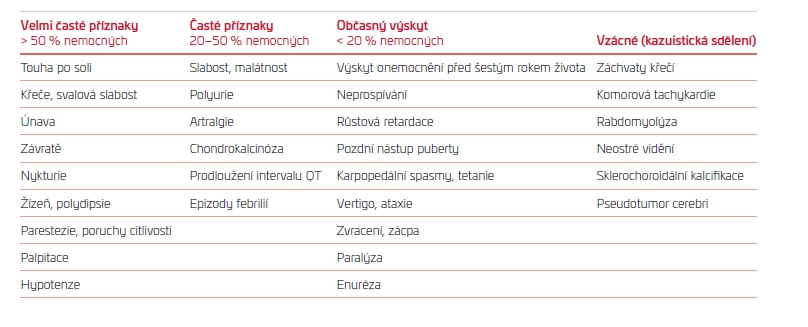
TAB. 1 Klinické manifestace a příznaky u pacientů s Gitelmanovým syndromem
TAB. 2 Diagnostická kritéria Gitelmanova syndromu

Protože povědomí o tomto onemocnění je stále poměrně malé a neexistovala shoda na tom, jak toto onemocnění diagnostikovat a pacienty léčit a vyšetřovat, zorganizovala pracovní skupina v rámci iniciativy KDIGO (Kidney Disease: Improving Global Outcomes) konferenci expertů zaměřenou na tuto problematiku. Výsledkem je komentovaná publikace, která shrnuje některé kontroverzní body a dává určitá doporučení pro klinickou praxi.
Typické projevy onemocnění a frekvence jejich výskytu jsou popsány v tab. 1. Diagnostická kritéria GS pak stručně shrnuje tab. 2.
Diferenciální diagnóza GS zahrnuje zejména klasickou formu Bartterova syndromu (BS, typ III). Hlavní odlišení od BS spočívá v nálezu hypokalciurie, hypomagnezemie (< 0,65 mmol/l) a hypermagneziurie s frakční exkrecí magnezia > 4 % u Gitelmanova syndromu. Rozlišení ale vždy musí přinést genetické potvrzení diagnózy. Dříve doporučovaný test s použitím hydrochlorothiazidu se dnes již nedoporučuje z důvodu jeho rizikovosti (možnost prohloubení deficitu iontů a dehydratace). Stejně tak se nedoporučuje provádět renální biopsii u malého počtu nemocných s GS, kteří mají proteinurii (bývá tubulárního charakteru při hypokalemické tubulopatii/nefropatii). Hypokalemická metabolická alkalóza s hypertenzí a zvýšenou kaliurií může být projevem primárního hyperaldosteronismu, Cushingova či Liddleova syndromu, kongenitální adrenální hyperplazie či stenózy renální tepny. Snížení koncentrace ionizovaného kalcia spolu s hypomagnezemií doprovází časné pooperační období po paratyreoidektomii, kdy ale většinou pozorujeme i hyperfosfatemii. Familiární hypomagnezemie s hyperkalciurií a nefrokalcinózou bývá způsobena mutací v genu CLDN16 pro claudin‑16 a ovlivňuje již zmíněný kanál TRPM6. Prognóza tohoto onemocnění je ale výrazně horší než u GS, a stav řady nemocných progreduje (zejména kvůli nefrokalcinóze) do stadia terminálního selhání ledvin. Odlišení těchto dvou chorob za situace, kdy není přítomna nefrokalcinóza, je možné prakticky jen geneticky.
Z dalších geneticky podmíněných chorob je možné zmínit mutace v genu HNF1B, kódujícím transkripční faktor HNF‑1β (HNF‑hepatocyte nuclear factor). Nemocní s mutacemi v tomto genu mohou imponovat jako pacienti s GS kvůli přítomnosti hypomagnezemie, ale další klinické projevy jsou odlišné (přítomnost MODY [maturity‑onset type diabetes of the young] diabetu, časné renální postižení s přítomností cyst, autozomálně dominantní typ dědičnosti, různé urogenitální malformace). Stejně tak syndrom EAST (epilepsie, ataxie, senzoricko‑neurální hluchota, tubulopatie) může být spojen s některými iontovými změnami podobně jako GS, ale má řadu extrarenálních projevů a je spojen s mutacemi v genu KCNJ10 (kódujícím kaliový kanál KCNJ10/Kir‑4). V diferenciální diagnóze je třeba vzít v úvahu i celou řadu dalších chorob, které vedou k tubulointersticiálnímu postižení (TI) a mohou způsobit iontové změny (např. Sjögrenův syndrom), a některé léky, jež blokují funkčnost kanálu NCCT.
Všichni nemocní splňující klinická a laboratorní kritéria pro GS by měli být geneticky testováni na přítomnost mutace v genu SLC12A3. Pokud nebude nalezena mutace (pravděpodobnost 20–30 %), je vhodné ještě doplnit genetickou analýzu o mutace v genu CLCNKB a HNF1B. Nové genetické techniky typu sekvenování nové generace (new generation sequencing, NGS) dnes již umožňují mnohem rychlejší diagnostiku a nabízejí i vyšetření určitého panelu mutací, které by se ke GS mohly vztahovat.
U pacientů s GS neomezujeme jejich potřebu soli, a naopak je nabádáme, aby solili dle potřeby. V léčbě GS pak je nutné suplementovat především ionty hořčíku, což rovněž vede ke kompenzaci močových ztrát chloridů a zlepšuje i odpověď na podávaný draslík. Z přípravků dáváme přednost chloridu hořečnatému či glycerofosfátu hořečnatému, dále pak organickým solím (aspartátu, citrátu, laktátu), které ale někdy mohou vyvolávat průjem. Celkovou denní dávku 300 mg (12,2 mmol Mg2+) rozdělujeme do dvou až tří denních porcí podávaných s jídlem. Trvalá suplementace hořčíku upravuje hypomagnezemii a je prevencí tetanie. Tím se rovněž snižuje deficit draslíku v organismu, i když běžně bývá nutné suplementovat i draslík. Podáváme ho zejména ve formě kalium chloridu, což částečně zmírňuje již tak významnou metabolickou alkalózu. Tablety nepodáváme na lačný žaludek, aby nedráždily žaludeční sliznici a byly lépe tolerovány; popřípadě je možné chlorid draselný (KCl) podat ve formě sirupu. Dávku titrujeme dle laboratorních hodnot. Intravenózní suplementace, zejména kalia, bývá nutná v případě poruchy srdečního rytmu, výskytu kvadruplegie, respiračního selhávání či rabdomyolýzy a někdy jako prevence arytmií v perioperačním období. Základem léčby chondrokalcinózy je suplementace magnezia. Při akutních symptomech s bolestmi pak často pomohou nesteroidní antiflogistika či kolchicin.
Komentář
- Prof. MUDr. Romana Ryšavá, CSc., Klinika nefrologie 1. LF UK a Všeobecné fakultní nemocnice v Praze
Literatura
- Cruz DN, Shaer AJ, Bia MJ, et al. Yale Gitelman’s and Bartter’s Syndrome Collaborative Study Group. Gitelman’s syndrome revisited: an evaluation of symptoms and health related quality of life. Kidney Int 2001;59:710–717.
- Graziani G, Fedeli C, Moroni L, et al. Gitelman syndrome: pathophysiological and clinical aspects. QJM 2010;103:741–748.
- Blanchard A, Vargas Poussou R, Vallet M, et al. Indomethacin, amiloride, or eplerenone for treating hypokalemia in Gitelman syndrome. J Am Soc Nephrol 2015;26:468–475.
- Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt losing tubulopathies. Am J Med 2002;112:183–190.
- Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008;3:22.
- Ryšavá R, Reiterová J, Urbanová M, et al. Gitelmanův syndrom jako častá příčina hypokalemie a hypomagnezemie. Vnitř Lék 2016;62(Suppl. 6):6S78–6S83.
- Kategorie: Komentované články
- Klíčová slova: Gitelmanův syndrom; hypokalemie
Komentovaná práce přináší velmi užitečný přehled o klinických projevech, možnostech diagnostiky a doporučeních pro léčbu nemocných s Gitelmanovým syndromem. Poukazuje také na úskalí, která jsou s diagnostikou a léčbou této choroby spojena.
Z klinické praxe dobře víme, že normalizace iontových změn je často velmi obtížná a že u většiny nemocných se musíme spokojit s kalemií kolem 3 mmol/l a magnezemií kolem 0,6 mmol/l; v řadě případů ale těchto hodnot nedosáhneme a snaha o navýšení suplementace léky vede k jejich horší toleranci s četnými nežádoucími projevy léčby (žaludeční ulcerace, zvracení, průjmy).
V některých studiích je popisován pozitivní vliv léků blokujících osu RAAS, které vedou ke zvýšení koncentrace draslíku (např. podáváním inhibitorů angiotenzin konvertujícího enzymu či antagonistů aldosteronu – spironolaktonu či eplerenonu). Snížení aktivace této osy může mít určitý kardioprotektivní účinek, ale bývá často spojeno s výskytem symptomatické hypotenze, zhoršením únavy a zvýšením ztrát chloridů do moči. U velmi těžkých forem onemocnění lze zkusit podávat i inhibitory cyklooxygenázy 1 (indometacin), které mohou částečně snížit vylučování sodíku, a tím následně i sekreci draslíku. Jejich vliv je však omezený, lépe fungují u neonatálních forem BS (u těch je, na rozdíl od GS, zvýšená močová sekrece prostaglandinu E2, kterou indometacin blokuje). Při této léčbě je navíc nutné pečlivě monitorovat případné nežádoucí účinky léku na trávicí ústrojí a také glomerulární filtraci. V nedávno publikované otevřené, randomizované studii se zkříženým uspořádáním se u 30 pacientů s GS srovnával vliv indometacinu (podávaného v jedné denní dávce 75 mg), eplerenonu (150 mg) a amiloridu (20 mg) na hypokalemii v průběhu šestitýdenní léčby.3 Léky byly podávány ke standardní suplementaci kalia a magnezia. Všechny léky vedly ke zvýšení sérové koncentrace kalia o cca 0,3 mmol/l. Zatímco amilorid a eplerenon vedly ke zvýšení odpadů sodíku, a tím k větší volumové depleci, indometacin byl spojen s poklesem glomerulární filtrace a u třetiny pacientů s gastrointestinální intoleran-cí. Všechny tři léky tedy měly podobný vliv na korekci hypokalemie, ale je nepochybné, že indometacin a další nesteroidní antiflogistika by měly být dlouhodobě podávány s velkou opatrností s ohledem na jejich nežádoucí účinky.
Odlišení od jiných tubulopatií se zvýšenými ztrátami soli (zejména BS) není vždy jednoduché. U malé části nemocných s GS byla prokázána přítomnost mutace v genu CLCNKB, který kóduje chloridový kanál ClC Kb, lokalizovaný jak v distálním stočeném kanálku (těsně vedle kanálu NCCT), tak také ve vzestupné části Henleovy kličky.4 To může způsobit, že tito jedinci mají jakýsi „překryvný“ syndrom mezi BS a GS (GS like fenotyp) s velmi variabilní fenotypovou manifestací (od neonatálního BS na jedné straně ke GS na straně druhé).5 Mutace v tomto genu by měly být vyšetřovány u všech nemocných s GS, zejména pak tam, kde nebyla prokázána mutace v genu SLC12A3, což po-tvrzují i naše pozorování.6 Je třeba také počítat s tím, že u 15–20 % pacientů s GS nalezneme kauzální mutaci jen na jedné alele, jelikož druhá se nejspíše nachází v regulačních oblastech genu (včetně intronů) anebo v jiných genech.
Projevy GS mohou být iatrogenně navozeny celou řadou léků, ale nejčastěji se s nimi setkáváme při chronickém abúzu diuretik thiazi-dového typu anebo laxativ (tab. 3). Při podezření na abúzus diuretik (zejména u mladých žen) je vhodné opakované vyšetření moči na stanovení jejich metabolitů. Laboratorně může pomoci zejména koncentrace vylučovaných chloridů; při GS jsou hodnoty chloridů v moči normální, nebo jen mírně zvýšené, zatímco diuretika zvyšují frakční exkreci chloridů několikanásobně nad normu. Pomoci může i test s intravenózně podaným furosemidem, který v případě GS nezvýší natriurézu ani kalciurii, zatímco při abúzu thiazidů se obojí signi-fikantně zvýší. Abúzus laxativ většinou bývá doprovázen metabolickou acidózou (ztrátami bikarbonátů stolicí) a poměr K+/kreatinin v moči bývá nižší než 1,5.
TAB. 3 Léky, jejichž podávání je nejčastěji provázeno možností rozvoje hypokalemie a hypomagnezemie