Erdheimova–Chesterova choroba: vzácné systémové onemocnění pohle-dem nefrologa – kazuistiky
Úvod
Erdheimova–Chesterova choroba (ECD) je vzácné histiocytární onemocnění s rozmanitými klinickými projevy, od indolentních, lokalizovaných forem až po život ohrožující multisystémové onemocnění. Má tendenci postihovat hlavně kosti, centrální nervový systém (CNS), retroperitoneum, endokrinní systém, srdce a velké cévy. Podle klasifikace Světové zdravotnické organizace (WHO) „Tumours of Haematopoietic and Lymphoid Tissues“ z roku 2016 je řazena mezi histiocytární neoplazie.1
Histiocyty jsou speciálním podtypem tkáňových makrofágů se schopností fagocytózy. Onemocnění je histologicky charakterizováno infiltrací tkání pěnitými buňkami, což jsou lipidem naplněné histiocyty s malými jádry.2 Etiologie ECD není zcela objasněna. Často jsou u pacientů detekovány somatické mutace vedoucí k aktivaci signální dráhy MAPK/ERK. Nejčastěji bývá popisována mutace genu BRAF (V600E), která se v současnosti využívá jako cíl tzv. cílené terapie, např. vemurafenibem (přípravek Zelboraf®).2 Diagnostika ECD je velmi obtížná, neboť projevy nemoci se u jednotlivých pacientů liší a jsou často spojeny se „zánětlivými“ laboratorními změnami, což může vést k mylné diagnóze. V této práci uvádíme dva případy pacientů s ECD, které jsme diagnostikovali na naší klinice.
Kazuistika I
80letý pacient s ischemickou chorobou srdeční, arteriální hypertenzí a diabetes mellitus 2. typu byl vyšetřován cestou spádové kardiologie pro dva měsíce trvající dušnost, suchý kašel, nechutenství a úbytek hmotnosti. Při rentgenovém (RTG) vyšetření hrudníku byl zjištěn bilaterální fluidothorax, ve výsledcích laboratorních vyšetření byla patrna mírná elevace hodnot C‑reaktivního proteinu (CRP) a mírné snížení renální funkce se sérovou koncentrací kreatininu 119 µmol/l. Po vyloučení kardiální etiologie byla pacientovi empiricky podána antibiotická terapie. Při opakovaných kontrolních vyšetřeních však RTG nález zůstával neměnný, stejně jako laboratorní parametry a klinický stav.
O šest měsíců později nemocný absolvoval pravidelné urologické vyšetření pro benigní hyperplazii prostaty. Vzhledem k nejasnému ultrasonografickému nálezu bylo indikováno vyšetření výpočetní tomografií (CT) urotraktu, které odhalilo bilaterální měkkotkáňové infiltrace v oblasti dutých systémů ledvin se stenózou vývodných cest a lehkým městnáním ledvin. Radiologický nález vzbudil podezření na autoimunitní etiologii či IgG4‑asociované onemocnění, pacient byl tedy odeslán k nefrologickému vyšetření a následně referován na naši kliniku.
Vzhledem k přetrvávajícímu bilaterálnímu fluidothoraxu byla provedena punkce pleurálního výpotku s nálezem exsudátu. Cytologické vyšetření prokázalo pouze řídkou lymfoplazmocytární celulizaci benigního charakteru, kultivační vyšetření bylo negativní. Následné vyšetření pozitronovou emisní tomografií v kombinaci s výpočetní tomografií (PET/CT) trupu popsalo pleurální výpotky do šíře až 60 mm, nevzdušnost přilehlého plicního parenchymu, oboustranné nepravidelné zesílení pleury a nepravidelný splývavý infiltrát v hilu obou ledvin a na mezenteriu. Po konzultaci s pneumology bylo přistoupeno k pleuroskopii s biopsií pleury, která však byla nevýtěžná z hlediska suspektní IgG4‑asociované choroby (fibrinózní exsudát, zánětlivý infiltrát, absence plazmatických buněk). Následovala laparoskopická biopsie radixu mezenteria s histologickým nálezem fibrohistiocytární léze nejisté biologické povahy, na jehož podkladě bylo vysloveno podezření na ECD či ALK‑pozitivní histiocytózu. Molekulárněgenetické vyšetření metodou PCR záhy potvrdilo mutaci v kodonu 600 genu BRAF typickou pro ECD.
Na doporučení hematologa byla doplněna série dalších vyšetření. Celotělové PET/CT zobrazilo kromě postižení pleury, ledvin a kořene mezenteria se zvýšenou akumulací 18F‑fluorodeoxyglukózy (FDG) nově také postižení distálních partií obou femurů (obr. 1), pravé fibuly a ložiskově sakra. Vyšetření kostní dřeně, magnetická rezonance (MR) mozku i echokardiografické vyšetření žádné abnormality neprokázaly.
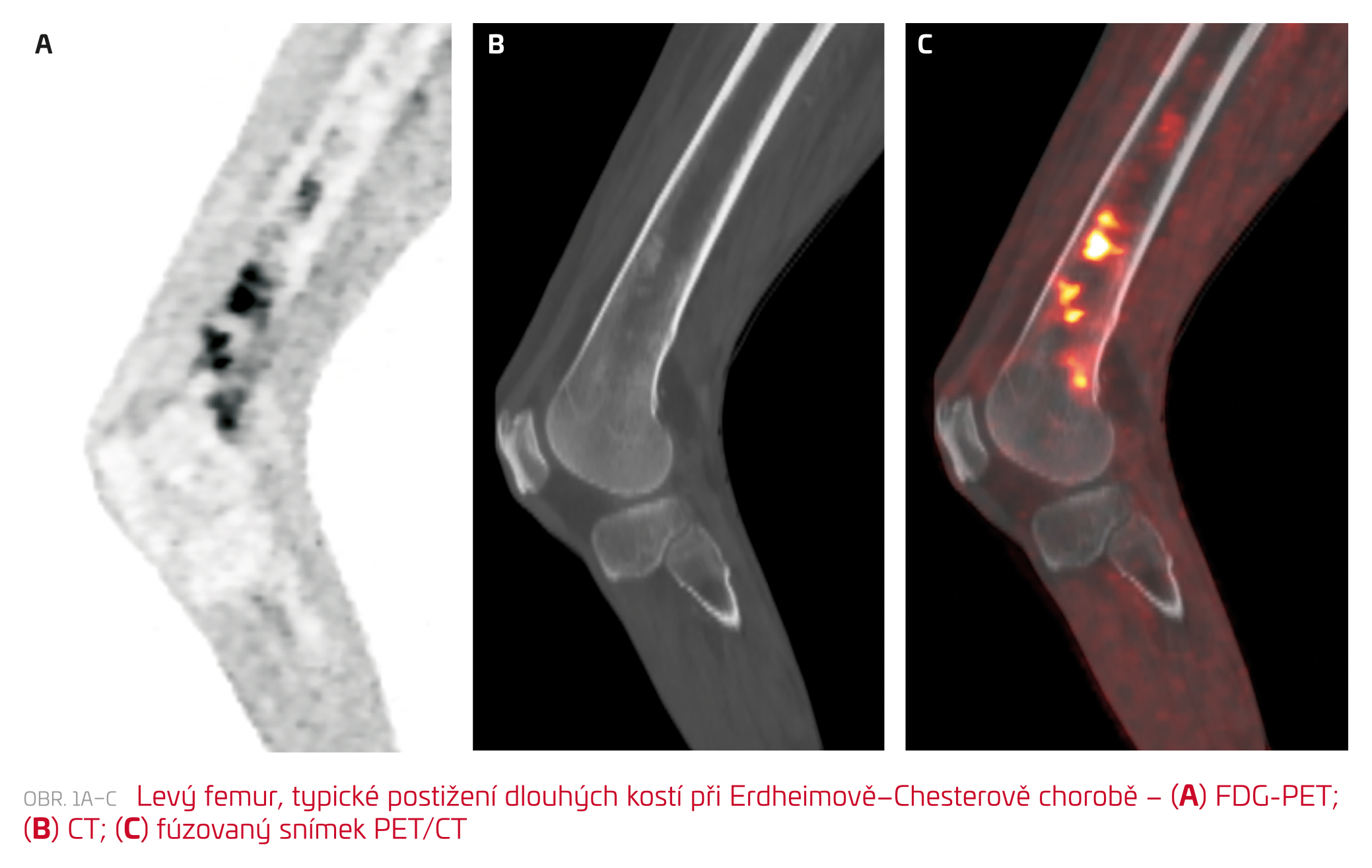
Terapie byla zahájena na hematologickém pracovišti podáváním kladribinu, cytotoxického purinového analoga inhibujícího syntézu a opravu DNA. Po schválení revizním lékařem byl pacient převeden na cílenou léčbu vemurafenibem, inhibitorem BRAF kinázy.
Kazuistika II
52letý pacient s hypertenzí, dyslipidemií, polytopním vertebrogenním algickým syndromem a anamnézou operace krční páteře byl praktickým lékařem odeslán na naši kliniku k diferenciálnědiagnostickému došetření dlouhodobých polytopních obtíží a patologického plicního nálezu. Pacient udával roky trvající suchý dráždivý kašel, zhoršující se při námaze. Na RTG hrudníku byl přítomen nespecifický nález, výpočetní tomografie s vysokým rozlišením (HRCT) plic prokázala difuzně ve všech lalocích rozložená ložiska typu opacit mléčného skla s diskrétní retikulací, bez lymfadenopatie, s mírnou progresí v čase.
Laboratorně byla zjištěna mírná renální insuficience se sérovou koncentrací kreatininu 116 µmol/l, smíšený močový nález s proteinurií 0,5 g/24 hodin a hyperkalciurií. Imunologické vyšetření prokázalo slabě pozitivní antinukleární protilátky (ANA), ostatní imunologické parametry byly negativní.
Byl konzultován pneumolog, který v diferenciální diagnostice zvažoval IgG4‑asociované onemocnění či non‑Langerhansovu histiocytózu a nevyloučil ani sarkoidózu. Nález doporučeného bronchoskopického vyšetření byl makroskopicky nevýtěžný, přítomna byla pouze lehká bronchitida bez známek vaskulitidy. Kultivační vyšetření bylo negativní. Následně bylo provedeno PET/CT vyšetření, které prokázalo větší infiltráty se zvýšenou akumulací FDG v obou plicích a dále sklerotická ložiska skeletu, místy rovněž se zvýšenou akumulací FDG.
Definitivní histologické vyšetření vzorku z bronchoskopie prokázalo histiocytózu, pro pozitivitu CD1a byl stav uzavřen jako histiocytóza z Langerhansových buněk. V dalším laboratorním došetření byla zjištěna i mírně zvýšená hodnota chitotriosidázy (128,9 nmol/h/ml), zatímco vyšetření angiotenzin konvertujícího enzymu (ACE) bylo v normě.
K posouzení možného postižení ledvin byla provedena renální biopsie, která neprokázala změny v souvislosti s histiocytózou ani s jiným systémovým onemocněním, přítomny byly vaskulární změny. Magnetická rezonance mozku zobrazila drobné nespecifické infiltráty v oblasti temporomandibulárních kloubů bilaterálně a infiltraci sfenoidální kosti, bez známek postižení mening či centrálního nervového systému (CNS). Radiologický obraz odpovídal ECD. Magnetická rezonance myokardu neprokázala postižení histiocytózou. Následně byla doplněna trepanobiopsie, jejíž nález byl v souladu s klinicky zvažovanou ECD. Histiocytóza z Langerhansových buněk v kostní dřeni prokázána nebyla. Molekulárněgenetickým vyšetřením byla jak v kostní dřeni, tak v plicní biopsii prokázána mutace v genu BRAF (V600E).
Pacient byl s diagnózou smíšené histiocytózy předán do péče hematologů a byla zahájena léčba cytarabinem.
Komentář
Literatura
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022;36:1703–1719.
- Řezáčová J, Kvasnička T, Dvořák P, et al. Erdheim‑Chester disease: radiologic findings of a rare multisystemic disease. Ces Radiol 2024;78:219–224.
- Goyal G, Heaney ML, Collin M, et al. Erdheim‑Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020;135:1929–1945.
- Sedrak P, Ketonen L, Hou P, et al. Erdheim‑Chester disease of the central nervous system: new manifestations of a rare disease. AJNR Am J Neuroradiol 2011;32:2126–2131.
- Starkebaum G, Hendrie P. Erdheim‑Chester disease. Best Pract Res Clin Rheumatol 2020;34:101510.
- Pegoraro F, Papo M, Maniscalco V, et al. Erdheim‑Chester disease: a rapidly evolving disease model. Leukemia 2020;34:2840–2857.
- Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim‑Chester disease but not in other non‑Langerhans cell histiocytosis. Blood 2012;120:2700–2703.
- Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600‑mutant Erdheim‑Chester disease and Langerhans cell histiocytosis: analysis of data from the histology‑independent, phase 2, open‑label VE‑BASKET study. JAMA Oncol 2018;4:384–388.
- Kategorie: Kazuistiky
- Klíčová slova: Erdheimova–Chesterova choroba
Erdheimova–Chesterova choroba je multisystémové onemocnění, typicky postihující více orgánů. Až 80 % pacientů má mutaci v genech signální dráhy mitogenem aktivované proteinkinázy (mitogen‑activated protein kinase, MAPK) / extracelulárně regulované kinázy (extracellular signal‑regulated kinase, ERK).3 Erdheimova–Chesterova choroba se nejčastěji objevuje u pacientů ve středním věku, častěji u mužů. V dětství je onemocnění velmi vzácné a postihuje především CNS.4
Projevy onemocnění jsou obecně velmi pestré a diagnostika bývá svízelná. Postižení CNS se projevuje mozečkovou symptomatologií, psychickými problémy či epilepsií. Dále může být přítomno oční postižení, například exoftalmus způsobený retrobulbární fibrózou. Časté jsou bolesti dlouhých kostí při postižení skeletu a současná přítomnost diabetes insipidus může vést k suspekci na ECD.4,5 Možné projevy postižení kardiovaskulárního aparátu jsou také velmi heterogenní, od perikarditidy po postižení myokardu; časté jsou i tumory v síních a komorách. Kolem velkých cév bývají na PET/CT patrny infiltráty, které mohou připomínat vaskulitidu velkých tepen (Takayasuovu arteritidu).5 Objevit se může i postižení plic, jater, sleziny, retroperitonea a kůže.
Pacient v první kazuistice byl opakovaně vyšetřován pro nespecifické příznaky, teploty, kašel a pleurální výpotky, opakovaně byl léčen antibiotickou terapií. Echokardiografie byla bez nálezu, neurologické vyšetření bylo v normě, punkce pleurálního výpotku byla nespecifická. Podezření na ECD bylo poprvé vysloveno na základě PET/CT vyšetření, kdy byl přítomen nález zesílení pleury, infiltrátů v hilu ledvin a infiltrátu v mezenteriu a zejména typický nález sklerotických změn v metafýzách femurů, fibuly a sakra. Diagnóza byla definitivně potvrzena histologickým vyšetřením infiltrátu v mezenteriu, kde byla prokázána fibrohistiocytární léze nejisté biologické povahy; následně bylo onemocnění klasifikováno jako ECD díky molekulárněgenetickému vyšetření, při němž byla prokázána mutace v kodonu 600 genu BRAF.
Ve druhé kazuistice se onemocnění u pacienta projevilo plicní symptomatologií; diferenciálnědiagnosticky byla zprvu zvažována sarkoidóza nebo ANCA‑asociovaná vaskulitida. Postižení ledvin, které může být též součástí ECD a nejčastěji se projevuje jako retroperitoneální fibróza s uzávěrem ureterů, u pacienta prokázáno nebylo. K diagnostice přispěl průkaz postižení kostí při vyšetření MR mozku, kde byl nález typický pro ECD, a to nespecifické infiltráty v oblasti temporomandibulárního skloubení a infiltrace sfenoidálních kostí. Definitivně byla diagnóza potvrzena biopsií z bronchoskopického vyšetření a trepanobiopsií, kde byla v obou případech prokázána mutace v genu BRAF. Pro pozitivitu CD1a v plicní biopsii nebylo možné zcela vyloučit ani smíšenou histiocytózu (ECD + histiocytóza z Langerhansových buněk).
Poslední doporučení pro diagnostiku a léčbu ECD bylo publikováno v roce 2020.3 Toto doporučení obsahuje podrobný výčet orgánového postižení a diferenciální diagnostiku, v níž jsou nejčastěji zvažována zánětlivá onemocnění, vaskulitidy, sarkoidóza či IgG4‑asociovaná onemocnění, která jsme zvažovali a vyloučili i v našich kazuistikách. Pro ECD je typické postižení skeletu, které lze prokázat celotělovou scintigrafií skeletu. V diagnostice je třeba provést vyšetření k potvrzení či vyvrácení orgánového postižení. Doporučeny jsou MR mozku, echokardiografie, neurologické vyšetření a PET/CT, kde je typický průkaz infiltrátů podél tepen. Dále mohou být přítomny retroperitoneální fibróza a postižení plic.6
Definitivní potvrzení nemoci přináší kombinace histologického vyšetření a průkazu genetické mutace.7 Problémem může být správný odběr biologického materiálu. V první kazuistice bylo vyšetření pleurálního výpotku i vyšetření pleury nepřínosné, až ve tkáni z mezenteria byla prokázána ECD. V druhém případě byl negativní nález v renální biopsii a obtížné bylo i zařazení nálezu z plicní biopsie. V transbronchiální biopsii plic byla stanovena diagnóza histiocytózy z Langerhansových buněk. Definitivní diagnóza ECD byla stanovena až histologickým vyšetřením kostní dřeně, kde nebyly prokázány buňky histiocytózy z Langerhansových buněk, ale pěnité makrofágy CD1a‑negativní a langerin‑negativní.
Onemocnění je léčeno hematoonkologem. Samostatná léčba kortikosteroidy je nedostatečná. V posledních letech se strategie léčby výrazně změnila díky možnostem ovlivnění alterované signální dráhy MAPK/ERK. K dispozici jsou inhibitory BRAF (vemurafenib, dabrafenib), inhibitory MEK (kobimetinib, trametinib) nebo kombinace inhibitorů BRAF a MEK. V první linii konvenční léčby se používají interferon alfa, kladribin či anakinra.
Jako druhá linie léčby jsou k dispozici infliximab, imatinib, sirolimus, tocilizumab a metotrexát.3,8
Závěr
Erdheimova–Chesterova choroba je velmi vzácné a obtížně diagnostikovatelné systémové onemocnění. Definitivní potvrzení diagnózy je založeno na histologickém a molekulárněgenetickém vyšetření. V léčbě jsou používány léky cílené na ovlivnění dráhy MAPK/ERK (inhibitory BRAF a inhibitory MEK), které mohou vést ke kompletní remisi onemocnění. Léčba však není bez nežádoucích účinků. Probíhají další klinické studie zaměřené především na kombinační léčbu inhibitory BRAF a MEK.