Bartterův a Gitelmanův syndrom
SOUHRN
Bartterův a Gitelmanův syndrom patří mezi tubulopatie spojené se solnými ztrátami. Tato vzácná onemocnění mohou být spojena se vznikem závažných elektrolytových abnormalit. Včasná diagnostika tubulopatií je klíčová pro zvolení adekvátní terapie. Pokroky v molekulární genetice umožnily identifikaci genů a patofyziologických mechanismů spojených se vznikem těchto chorob. Náš přehledový článek pojednává o etiologii a diagnostice těchto onemocnění z pohledu současných poznatků. Zároveň předkládáme aktuální léčebná doporučení.
Úvod
Správná funkce lidského těla je závislá na vodní a elektrolytové rovnováze. Hlavním orgánem zodpovědným za udržení této rovnováhy jsou ledviny. Mechanismem glomerulární filtrace ledviny dospělého člověka za jediný den profiltrují okolo 180 litrů plazmy, jež obsahuje značné množství organických i anorganických látek, které jsou z velké části následně zpětně resorbovány systémem renálních tubulů. Okolo 25 000 mmol sodíku, 4 500 mmol bikarbonátu, 720 mmol draslíku a 900 mmol glukózy se denně dostává do tubulárního systému, jehož úkolem je navrátit tyto látky zpět do krevního oběhu. Ledviny proto představují energeticky nejnáročnější orgán v lidském těle. Pokud dojde k poruše reabsorpce elektrolytů kdekoliv v průběhu tubulárního systému, naruší se homeostáza a dojde k projevům onemocnění. V tomto přehledovém článku se věnujeme dvěma vrozeným onemocněním, která jsou spojena s poruchou zpětného vstřebávání elektrolytů. Na počátku 60. let 20. století popsali lékaři Pacita Pronove a Frederic Bartter nový syndrom charakterizovaný hypertrofií juxtaglomerulárního aparátu a hyperaldosteronismem u dvou pacientů s normálním krevním tlakem. Tato choroba dostala následně název Bartterův syndrom (BS).1 O čtyři roky později publikoval Hilel Gitelman kazuistiku dvou sester s novým onemocněním, které bylo charakterizováno hypokalemií a hypomagnezemií a po svém objeviteli pojmenováno Gitelmanův syndrom (GS).2 BS i GS jsou tubulopatie spojené se solnými ztrátami, proto je pro ně typický kompenzatorní hyperaldosteronismus vedoucí ke zpětné resorpci natria na úkor ztrát kalia ve sběrném kanálku. Pozdější výzkum prokázal, že obě tato onemocnění jsou dědičná autozomálně recesivně a mají velmi nízkou prevalenci – 1 : 100 000 u BS a 25 : 100 000 u GS. Zatímco genový defekt ústící v postižení transportních mechanismů v tlusté části vzestupného raménka Henleovy kličky je zodpovědný za vznik BS, u pacientů s GS je příčinou porucha funkce iontového kanálu v distálním tubulu.3
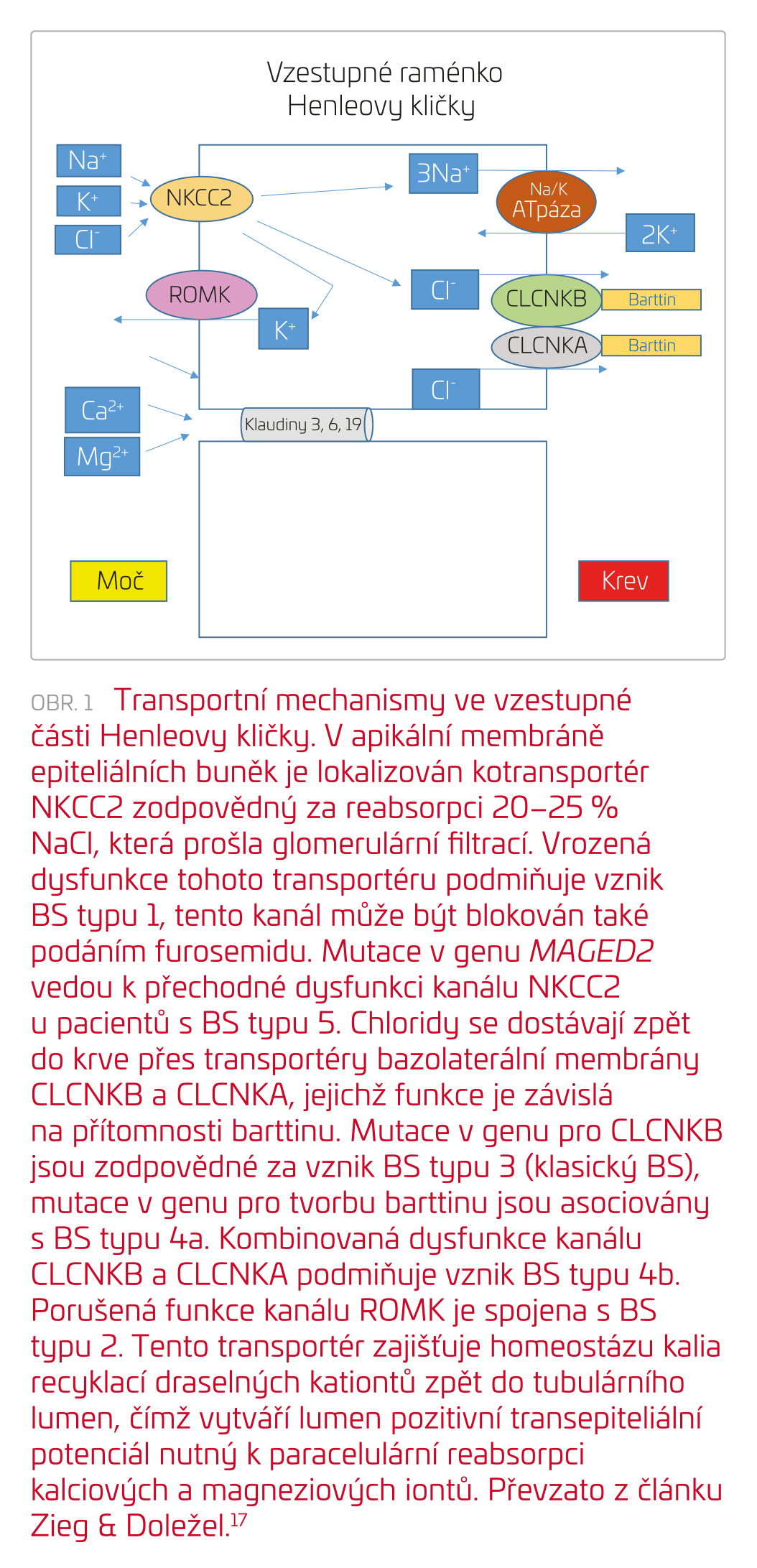
Bartterův syndrom
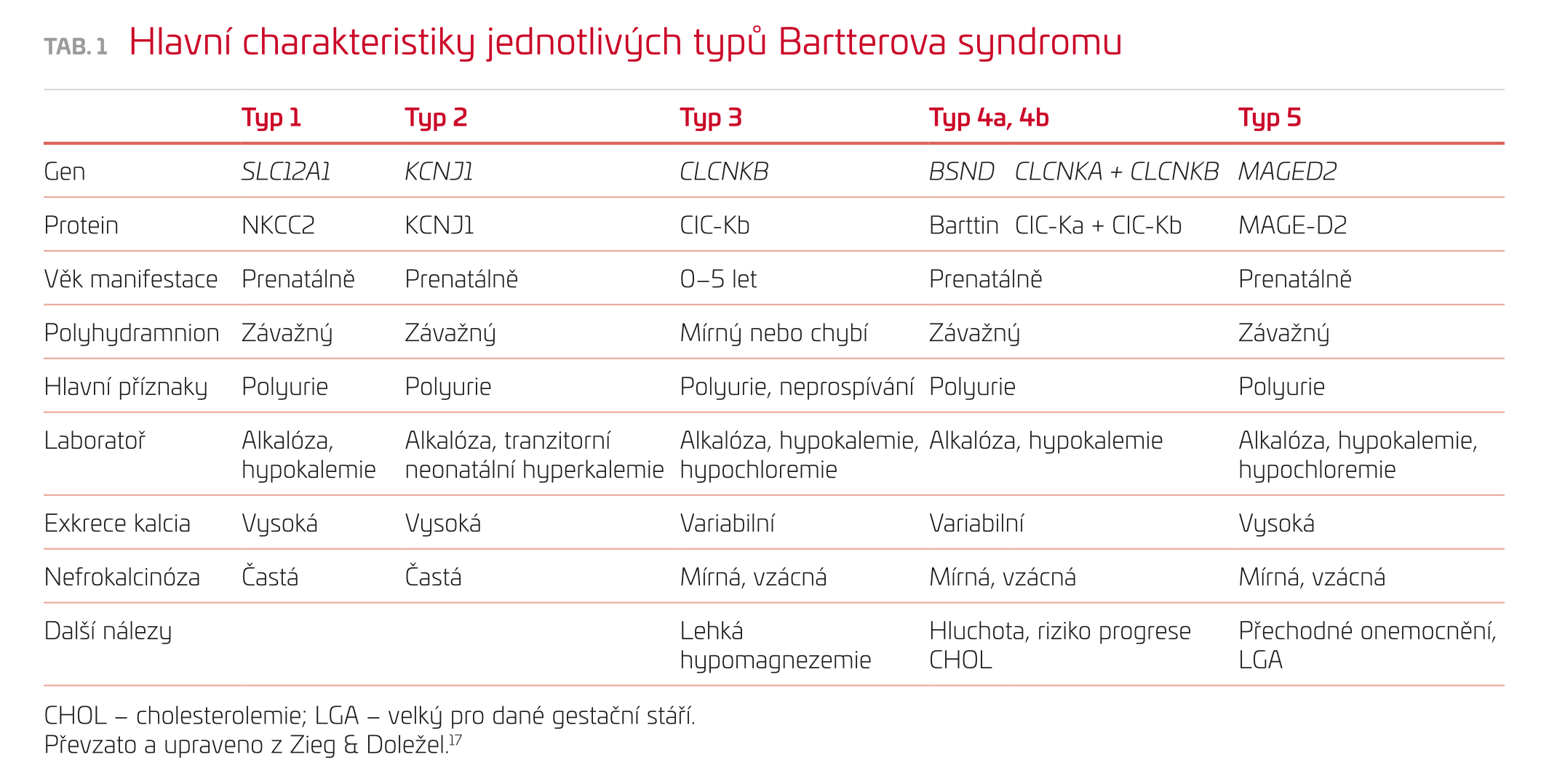
Bartterův syndrom (BS) představuje skupinu onemocnění charakterizovanou polyurií, hypokalemií, metabolickou alkalózou, hyperreninovým hyperaldosteronismem a normotenzí. V současné době rozeznáváme pět typů Bartterova syndromu v závislosti na genovém defektu. Na toto onemocnění často pomýšlíme již v prenatálním období, neboť u většiny matek bývá přítomen polyhydramnion. Pro jedince s BS je dále typické růstové neprospívání, u některých typů BS diagnostikujeme hyperkalciurii a nefrokalcinózu (tab. 1). Základním patofyziologickým mechanismem BS je porucha funkce tlusté části vzestupného raménka Henleovy kličky, která představuje zásadní komponentu mechanismu koncentrační schopnosti nefronu i transportu elektrolytů.4 Následkem porušené funkce této části nefronu dochází k rozvoji polyurie, dehydratace a ke vzniku polyhdramnionu. Solné ztráty vedou k aktivaci osy renin‑angiotenzin‑aldosteron (RAA) s následným zvýšeným vylučováním kalia a chloridů, v laboratoři nacházíme známky hypochloremické metabolické alkalózy s hypokalemií. Zároveň dochází k porušení mechanismu tubuloglomerulárního feedbacku s aktivací cyklooxygenázy 2, macula densa detekuje sníženou koncentraci chloridových iontů v tubulárním filtrátu, následkem čehož dochází k produkci velkého množství prostaglandinů, jež také stimulují produkci reninu a aktivují osu RAA s cílem udržet intravaskulární objem.5 Porucha reabsorpce solí ve vzestupném raménku Henleovy kličky má za následek sníženou resorpci kalcia vedoucí k hyperkalciurii a progresivní medulární nefrokalcinóze, a dále k narušení osmotického gradientu ve dřeni ledvin, a tím k poruše koncentrační schopnosti ledvin. Elektrolytový transport ve vzestupném raménku Henleovy kličky znázorňuje obrázek 1. Přenos onemocnění je autozomálně recesivní, pouze BS typ 5 se přenáší gonozomálně recesivně (tab. 1).6,7
BS typy 1, 2, 4 a 5 se klasicky manifestují již před narozením polyhydramnionem matky, který se objevuje mezi 20. a 30. týdnem gravidity.8 Po porodu, který je obvykle předčasný, pozorujeme rychlý hmotnostní úbytek při polyurii, v laboratorních výsledcích nacházíme metabolickou alkalózu s hypokalemií, vysoké hodnoty reninu a aldosteronu. Typická pro BS je dále zvýšená frakční exkrece chloridů (FECl) > 0,5 %.9 U části pacientů zjišťujeme hyperkalciurii, příp. nefrokalcinózu. Jedinci s BS typu 2 mají často v novorozeneckém období přechodnou acidózu a hyperkalemii.10 U většiny pacientů se BS typu 3 obvykle manifestuje ve věku po prvním roce života neprospíváním a polyurií, ale i u tohoto typu BS byla již popsána prenatální manifestace.11 Mezi příznaky mladších dětí s manifestací BS typu 3 patří žízeň, obstipace, febrilie, hypotonie či opakované zvracení. V pozdějším věku se objevuje častěji svalová slabost, únava, v prepubertálním období je obvykle patrné růstové neprospívání. BS typu 4 je spojen s percepční nedoslýchavostí. Pro děti s BS je také typická preference slaných potravin. I když můžeme často vyjádřit suspekci na BS na základě anamnézy, laboratorních a zobrazovacích vyšetření, je v současnosti k potvrzení diagnózy vzhledem ke značné fenotypové variabilitě doporučeno provést molekulárněgenetické vyšetření.3,6,12
Gitelmanův syndrom
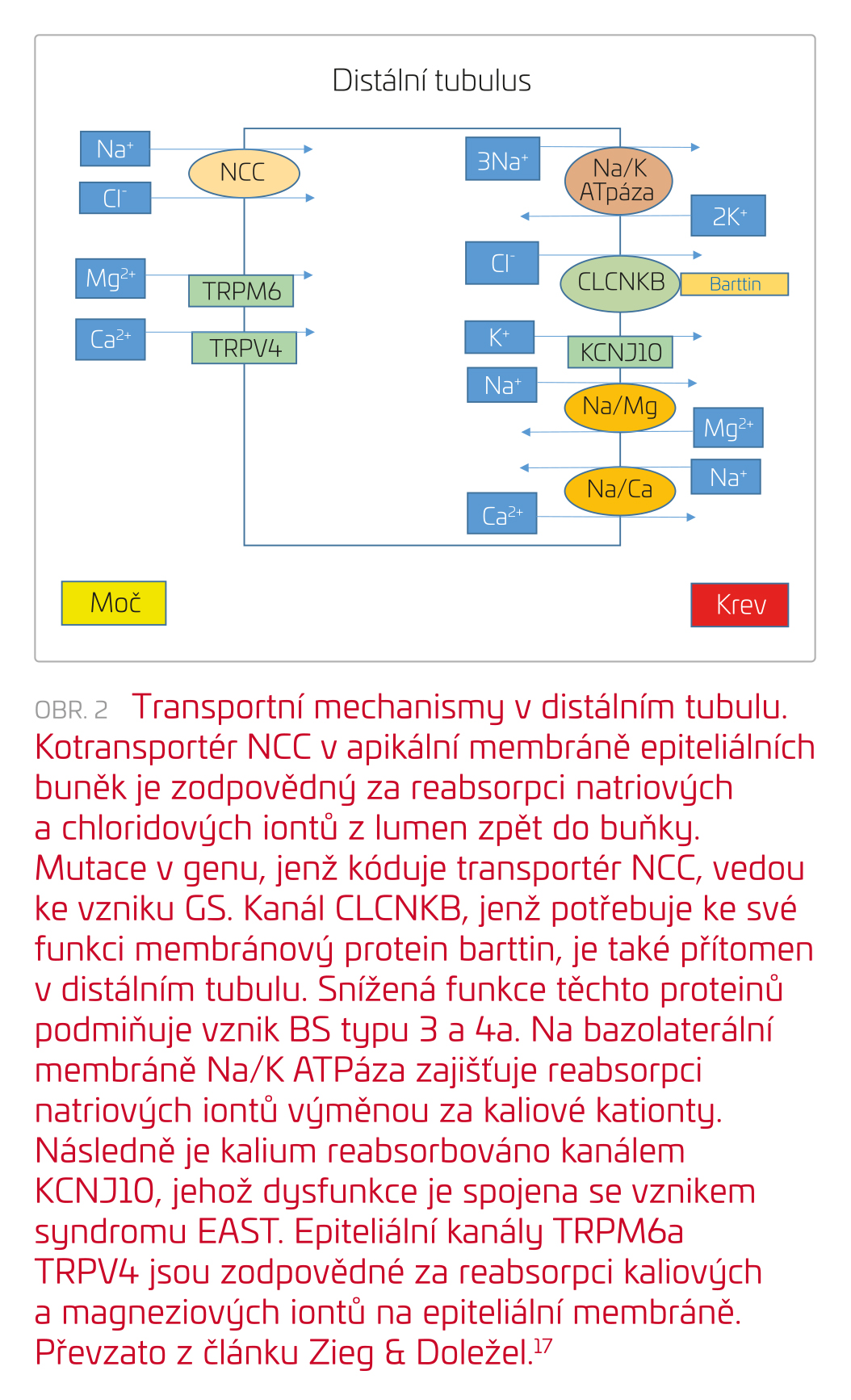
Gitelmanův syndrom (GS) je podmíněn solnými ztrátami spojenými s poruchou funkce thiazid‑senzitivního symportéru NaCl v apikální membráně distálního tubulu, tento kanál je kódován genem SLC12A3.13 Transportní mechanismy v distálním tubulu znázorňuje obrázek 2. Mezi hlavní projevy GS patří také hypokalemie, metabolická alkalóza jako důsledek aktivace osy RAA, typický je však nález hypomagnezemie a hypokalciurie. Taktéž pacienti s GS jsou normotenzní.14 Hypomagnezemie je způsobena zvýšenými ztrátami hořčíku do moči na podkladě snížené exprese luminálního kanálu TRPM6 v distálním tubulu, hypokalciurie vzniká následkem zvýšené pasivní reabsorpce kalciových iontů v proximálním tubulu.7 Fenotyp GS může být přítomen i u pacientů s BS typu 3. GS se obvykle projevuje v adolescenci či dospělosti, ale byly popsány i případy manifestace v neonatálním období.15 Mezi hlavní příznaky patří chuť na slané potraviny, polydipsie, nykturie, svalová slabost, únava, fyzická nevýkonnost, tetanie, parestezie, palpitace a hypotenze. U dětí je často patrné opoždění růstu a puberty.16
Diagnostika
V rámci prenatální diagnostiky pomýšlíme na BS při nálezu časného polyhydramnionu matky. Je možné využít prenatálně molekulárněgenetického vyšetření, v případě jeho nedostupnosti lze zvážit biochemické vyšetření plodové vody, jehož výsledky k odlišení BS od jiných příčin polyhydramnionu ale nejsou dle současných poznatků zcela spolehlivé.18
U dítěte již narozeného je zásadní anamnéza polyhydramnionu matky, dále anamnéza polyurie, dehydratace, horeček, neprospívání či dalších symptomů BS. Posouzení iontogramu a acidobazické rovnováhy je zásadní, definitivní diagnózu potvrdí molekulárněgenetické vyšetření. Prvním příznakem po porodu je hypovolemie v důsledku močových ztrát vody a solí. Laboratorní změny nemusejí být v prvních dnech života plně vyjádřeny. U jedinců s BS typu 2 se může přechodně v novorozeneckém období vyskytnout hyperkalemie doprovázená metabolickou acidózou, u pacientů s BS typu 3 bývá v laboratorních nálezech přítomna hypomagnezemie.6
Diferenciální diagnostika
V diferenciální diagnóze BS a GS musíme zvažovat kongenitální chloridový průjem spojený se sníženou resorpcí chloridů v ileu a kolon. Toto onemocnění se projevuje vodnatými průjmy s vysokým obsahem chloridových iontů.19 Sonograficky je patrné rozšíření lumen střevních kliček. Na rozdíl od BS u těchto pacientů nacházíme nízké odpady chloridů do moči a nízkou FECl.9 Nízká FECl je charakteristická pro tzv. pseudo‑Bartterův syndrom (PBS), jenž zahrnuje skupinu získaných onemocnění extrarenálního původu, která mohou činit diferenciálnědiagnostické obtíže, jelikož jsou charakterizována obdobným nálezem v iontogramu a ve vyšetření acidobazické rovnováhy jako tubulopatie se solnými ztrátami. Mezi nejčastější příčiny PBS patří zneužívání diuretik, laxativ a opakované zvracení. Může se objevit i při užívání některých aminoglykosidových antibiotik (gentamicin, kolistin) či u pacientů s cystickou fibrózou, Pendredovým syndromem a u jedinců s omezením chloridů ve stravě.20 S abúzem diuretik, laxativ a vyvolávaným zvracením se nejčastěji setkáváme u dívek a žen s poruchou příjmu potravy či jinými psychopatologiemi. Močové koncentrace natria, kalia a chloridů bývají zvýšené časně po podání diuretik, k jejich snížení poté dochází s odstupem času od užití léků. Diagnostické je proto testování moči na přítomnost diuretik spolu s frakční exkrecí chloridů. Pozitivní screening moči na přítomnost diuretik spolu s FECl > 0,5 % podporuje zneužívání diuretik jako příčinu PBS. Zneužívání laxativ je spojeno s hypokalemií vzhledem k vysokému obsahu kalia v dolní etáži gastrointestinálního traktu. Tito pacienti mívají typicky nízké koncentrace natria, chloridů a vysoké koncentrace kalia v moči při sekundárním hyperaldosteronismu.21 U pacientů s vyprovokovaným zvracením vídáme metabolickou alkalózu a hypokalemii z renálních ztrát v důsledku sekundárního hyperaldosteronismu.22 PBS spojený se zneužíváním laxativ a diuretik představuje rizikový faktor rozvoje chronického onemocnění ledvin.21 Také HNF1B nefropatie se může manifestovat fenotypem GS, pro toto onemocnění je ale typický nález renálních cyst, vrozených vad vývodného močového systému a rozvoj diabetu MODY5.23 Hypokalemií, hypomagnezemií a metabolickou alkalózou se projevuje také syndrom epilepsie, ataxie, percepční nedoslýchavosti, tubulopatie (EAST), mezi jehož příznaky ale navíc patří ataxie, epilepsie, opoždění psychomotorického vývoje a percepční hluchota.6,24 U pacientů s hypokalemickou metabolickou alkalózou a hypertenzí je nutno zvažovat jiné diagnózy, jako je např. primární hyperaldosteronismus, Liddleův syndrom nebo syndrom zdánlivého nadbytku mineralokortikoidů.
Léčba Bartterova syndromu
Terapie je započata bez znalosti výsledku genetického vyšetření s cílem předejít rozvratu vnitřního prostředí.12 Recentně byla publikována nová doporučení pro léčbu BS i GS u dětí.6,16 Základem léčby BS je úprava diety a substituce elektrolytů, kterou je vhodné rozdělit alespoň do tří denních dávek (ideálně více dle tolerance pacienta) vzhledem ke kontinuálním ztrátám iontů a vody do moči. Hypokalemie je závažným projevem BS, významný pokles koncentrace kalia v séru může pacienta ohrozit hlavně fatálními arytmiemi.9 Kalium obvykle hradíme ve formě chloridu draselného (KCl) s cílem dosáhnout koncentrace kalia v séru alespoň 3 mmol/l. KCl nepodáváme nalačno, abychom dosáhli lepší tolerance. Jiné sloučeniny kalia, jako je např. citrát, nejsou k substituci vhodné vzhledem k možnému zhoršení alkalózy. V případě hypomagnezemie podáváme soli hořčíku, aspartát, citrát a laktát mají lepší biologickou dostupnost v porovnání s anorganickými sloučeninami (oxid, hydroxid).25 Cílová koncentrace magnezia v séru je > 0,6 mmol/l. V léčbě se dále užívají nesteroidní protizánětlivé léky, inhibitory cyklooxygenázy (COX), které snižují produkci prostaglandinů, zmírňují symptomy a příznivě ovlivňují iontogram, glomerulární filtraci i růst dětí s BS. V současnosti nemáme dostatek dat k určení inhibitoru COX první volby. V případě jejich podávání je nutno vzít v potaz možný rozvoj gastrointestinálních nežádoucích účinků, při podávání neselektivních inhibitorů COX se doporučuje současné užívání inhibitorů protonové pumpy s vědomím rizika vzniku hypomagnezemie. Terapii inhibitory COX nasazujeme u euvolemických pacientů s vědomím rizika možné nefrotoxicity. Seznam inhibitorů COX užívaných v léčbě BS shrnuje tabulka 2. V současnosti není pacientům s BS podávání kalium šetřících diuretik, thiazidů a léků inhibujících osu RAA obecně doporučováno vzhledem k riziku vzniku hypovolemie a potenciace solných ztrát. Jejich případné nasazení je namístě u jedinců s vážnými iontovými abnormalitami, které přetrvávají při maximální intenzitě léčby substitucí kalia a nesteroidními protizánětlivými léky.6 U některých pacientů s BS a růstovým neprospíváním lze při nedostatečné produkci růstového hormonu zvážit zahájení léčby růstovým hormonem, ale to až po adekvátní metabolické kompenzaci základního onemocnění výše zmíněnou konzervativní léčbou.26

Léčba Gitelmanova syndromu
Suplementace elektrolytů je hlavní léčebnou modalitou taktéž v případě GS. Doporučuje se neomezený příjem soli, který obvykle dostatečně pokryje potřebu pacienta. Iniciální dávka elementárního magnezia je 5 mg/kg, 0,2 mmol/kg na den ve 2–4 denních dávkách. Iontová suplementace se podává ideálně s jídlem. Navyšování dávky magnezia je někdy limitováno rozvojem gastrointestinálních komplikací (průjem). V případě závažné hypomagnezemie s rozvojem tetanie je vhodné podat magnezium intravenózně – MgSO4 25–50 mg/kg, což odpovídá 0,1–0,2 mmol/kg elementárního Mg/kg v jedné dávce. Maximální dávka představuje 2 g MgSO4 v jedné infuzi, tuto substituci lze opakovat v intervalu šesti hodin.13,27 10 ml 10% MgSO4 obsahuje 1 g MgSO4, 98,6 mg a 4 mmol elementárního magnezia. Kalium je vhodné substituovat primárně perorálně ve formě KCl v několika denních dávkách. Intravenózní podání KCl je vyhrazeno pro jedince, kteří nemohou přijímat perorální substituci, nebo pro pacienty s významnou hypokalemií. KCl se podává ředěný v solném roztoku na koncentraci 40 mmol/l, rychlost podání infuze do periferie by neměla překročil 10 mmol/h, protože vyšší koncentrace mohou iritovat cévní endotel. V případě přetrvávající symptomatické hypokalemie, kterou nelze dostatečně hradit perorální substitucí, je možno dítěti s GS podat léky inhibující osu RAA, kalium šetřící diuretika nebo nesteroidní protizánětlivé léky, případně jejich kombinace. Podávání spironolaktonu může být spojeno s antiandrogenními nežádoucími účinky (gynekomastie, erektilní dysfunkce, nepravidelnost menstruačního cyklu), proto dáváme přednost nasazení eplerenonu, selektivního antagonisty aldosteronu.28 Stejně jako v případě BS musíme brát v potaz rizika podávání diuretik vzhledem k možné potenciaci solných ztrát a hypovolemie. Při dehydrataci (např. zvracení, průjmy) je nutné léčbu těmito přípravky dočasně přerušit. Indikace podávání inhibitorů COX u jedinců s GS je sporná, protože močové koncentrace prostaglandinu E2 nebývají obvykle zvýšeny. U pacientů s tubulopatiemi se solnými ztrátami je nutné některé léky buď zcela vynechat, nebo je podávat se zvýšenou opatrností. Jedná se o přípravky, které mohou potencovat hypokalemii nebo hypomagnezemii, jako jsou např. některá diuretika či laxativa. I pacienti s GS jsou léčeni růstovým hormonem v případě, že mají kompenzované onemocnění a splní indikační kritéria.
Prognóza
U významné části pacientů s BS v průběhu sledování dochází k poruše glomerulární filtrace. Příčina zhoršování funkce ledvin není zatím jasná. Může se na ní podílet chronická hypokalemie, dlouhodobě zvýšené koncentrace prostaglandinů či aldosteronu v krvi. Vliv léčby inhibitory COX či nefrokalcinózy na progresi renálního selhání nebyl doposud potvrzen.3,29 Rozvoj chronického onemocnění ledvin spojeného s poklesem funkce ledvin zjišťujeme u pacientů s GS výrazně méně často. Nebyl také popsán vliv GS na dobu dožití. Obecně je vhodné děti s BS a GS pravidelně sledovat v ambulanci dětské či dospělé nefrologie v rámci center pro vzácná onemocnění.
Literatura
- Bartter FC, Pronove P, Gill JR, Jr., Maccardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med 1962;33:811–828.
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 1966;79:221–235.
- Walsh PR, Tse Y, Ashton E, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J 2018;11:302–309.
- Kleta R, Bockenhauer D. Bartter syndromes and other salt‑losing tubulopathies. Nephron Physiol 2006;104:73–80.
- Reinalter SC, Jeck N, Brochhausen C, et al. Role of cyclooxygenase‑2 in hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int 2002;62:253–260.
- Konrad M, Nijenhuis T, Ariceta G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int 2021;99:324–335.
- Besouw MTP, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: Questions of class. Pediatr Nephrol 2020;35:1815–1824.
- Laghmani K, Beck BB, Yang SS, et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and MAGED2 Mutations. N Engl J Med 2016;374:1853–1863.
- Zieg J, Gonsorcikova L, Landau D. Current views on the diagnosis and management of hypokalaemia in children. Acta Paediatr 2016;105:762–772.
- Finer G, Shalev H, Birk OS, et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr 2003;142:318–323.
- Yang X, Zhang G, Wang M, et al. Bartter Syndrome Type 3: Phenotype‑Genotype Correlation and Favorable Response to Ibuprofen. Front Pediatr 2018;6:153.
- Doležel Z, Holeszová A, Ráčilová Z, et al. Bartterův syndrom – klinicko‑genetická analýza. Pediatr praxi 2015;16:179–182.
- Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008;3:22.
- Doležel Z, Jeřábek M. Gitelmanův syndrom. Pediatr praxi 2021;22: 216–217.
- Tammaro F, Bettinelli A, Cattarelli D, et al. Early appearance of hypokalemia in Gitelman syndrome. Pediatr Nephrol 2010;25:2179–2182.
- Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2017;91:24–33.
- Zieg J, Dolezel Z. Bartter and Gitelman syndromes. Čas Lék čes 2022;161:131–134.
- Allaf B, Dreux S, Schmitz T, et al. Amniotic fluid biochemistry in isolated polyhydramnios: a series of 464 cases. Prenat Diagn 2015;35:1331–1335.
- Di Meglio L, Castaldo G, Mosca C, et al. Congenital chloride diarrhea clinical features and management: a systematic review. Pediatr Res 2021;90:23–29.
- Najafi M, Kordi‑Tamandani DM, Behjati F, et al. Mimicry and well known genetic friends: molecular diagnosis in an Iranian cohort of suspected Bartter syndrome and proposition of an algorithm for clinical differential diagnosis. Orphanet J Rare Dis 2019;14:41.
- Jdiaa SS, Walsh SB, Bockenhauer D, et al. The hypokalemia mystery: distinguishing Gitelman and Bartter syndromes from ‘pseudo‑Bartter syndrome’. Nephrol Dial Transplant 2021;37:29–30.
- Do C, Vasquez PC, Soleimani M. Metabolic Alkalosis Pathogenesis, Diagnosis, and Treatment: Core Curriculum 2022. Am J Kidney Dis 2022;80:536–551.
- Adalat S, Hayes WN, Bryant WA, et al. HNF1B Mutations Are Associated With a Gitelman‑like Tubulopathy That Develops During Childhood. Kidney Int Rep 2019;4:1304–1311.
- Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 2009;360:1960–1970.
- Ranade VV, Somberg JC. Bioavailability and pharmacokinetics of magnesium after administration of magnesium salts to humans. Am J Ther 2001;8:345–357.
- Buyukcelik M, Keskin M, Kilic BD, et al. Bartter syndrome and growth hormone deficiency: three cases. Pediatr Nephrol 2012;27:2145–2148.
- Kraft MD, Btaiche IF, Sacks GS, Kudsk KA. Treatment of electrolyte disorders in adult patients in the intensive care unit. Am J Health Syst Pharm 2005;62:1663–1682.
- Morton A. Eplerenone in the treatment of Gitelman’s syndrome. Intern Med J 2008;38:377.
- Reinalter SC, Grone HJ, Konrad M, et al. Evaluation of long‑term treatment with indomethacin in hereditary hypokalemic salt‑losing tubulopathies. J Pediatr 2001;139:398–406.
- Kategorie: Přehledové články
- Klíčová slova: Bartterův syndrom; Gitelmanův syndrom; léčba; tubulopatie