Postižení plic u systémových nemocí pojiva
Souhrn
Systémové nemoci postihují řadu tkání a orgánů, a tito nemocní se tedy dostávají do péče odborníků z různých oborů medicíny (revmatologie, nefrologie, neurologie, kardiologie, pneumologie). Většina z těchto nemocí ve svém průběhu může postihovat i plicní tkáň. Fenotypy postižení plicní tkáně jsou nespecifické pro danou systémovou nemoc pojiva (SNP) a obvykle kopírují fenotypy idiopatických intersticiálních pneumonií. V rámci jedné SNP se může vyskytnout několik různých fenotypů plicního postižení. Jednoznačné rozklíčování fenotypu je nutné pro určení biologické povahy a závažnosti plicního postižení, a tím i pro jeho léčbu. Zánětlivé typy postižení léčíme protizánětlivou léčbou, typy fibrotické pak při progresivním charakteru fibrózy léčbou antifibrotickou. V tomto článku se budu věnovat pouze SNP bez systémových vaskulitid, které mají poněkud jiný imunopatologický mechanismus a ve většině případů i jiné typy postižení plic.
Úvod
Systémové nemoci pojiva (SNP) jsou charakterizovány autoimunitním procesem s tvorbou autoprotilátek zaměřených proti různým buněčným epitopům. Postihují řadu tkání a orgánů, z nichž časté je i postižení plic a ledvin. Nemocní jsou obvykle sledováni a léčeni revmatologem, nicméně v případě orgánových postižení je nutná spolupráce s dalšími specialisty: nefrology, pneumology, kardiology a neurology. V případě plicního postižení je někdy obtížné diferenciálnědiagnosticky odlišit, zda se jedná o intersticiální plicní proces (IPP) v rámci SNP, nebo zda je obraz dán polékovým postižením při léčbě SNP, nebo difuzní infekční pneumonií v imunokompromitovaném terénu u nemocného léčeného imunosupresivy. Plíce mohou být postiženy samostatně, v některých případech však může být postižena i pleura, klouby a svaly hrudního koše, případně plicní vaskulatura (tab. 1). V případě intersticiálního plicního postižení jednotlivé fenotypy de facto kopírují obraz idiopatických intersticiálních pneumonií a bez obrazu systémového postižení jsou od nich klinicko‑radiologicky neodlišitelné (tab. 2). Někdy může být plicní postižení jediným manifestním postižením u jinak klinicky inaparentně probíhající SNP. Je proto třeba pátrat i po minimálních projevech mimoplicního postižení a pozitivitě autoprotilátek. Je nutné si uvědomit, že asi 15 % původně IPP představují vlastní frustní formy SNP. Pokud jsou projevy nemoci omezeny pouze na plíce a jsou přítomny autoprotilátky bez jakýchkoliv mimoplicních projevů SNP, někteří autoři nově takové onemocnění označují jako SNP s plicní dominancí (lung‑dominant connective tissue disease – LD‑CTD). Vzhledem k četnosti plicního postižení u SNP je u některých SNP doporučován screening plicního postižení.1–3

Epidemiologie
Nejvyšší výskyt IPP (70 %) je u systémové sklerodermie (SSc), kde je fibrotický IPP také nejčastější příčinou úmrtí. V případě revmatoidní artritidy (RA) se vyskytuje plicní postižení u přibližně 20 % pacientů a nebývá obvykle klinicky závažné, při idiopatických zánětlivých myopatiích postihuje IPP 30 % pacientů. U systémového lupusu erythematodes (SLE) je klinicky významné postižení popisováno v 5 %, nicméně subklinické formy IPP jsou nalezeny u 30 % pacientů. Sjögrenův syndrom (SS) je provázen plicním postižením u 30 % pacientů, jen u 10 % je však klinicky významné. U ankylozující spondylitidy (AS) bývají intersticiální změny minimální a nespecifické, klinicky významné onemocnění ve formě bulózního emfyzému v horních lalocích se objevuje zřídka.1.2,4
Etiopatogeneze
U SNP se pravděpodobně podílejí na tkáňovém a pravděpodobně i na plicním postižení autoprotilátky, vyvoláním zánětlivého procesu a procesu hojení s poruchou architektoniky tkání. Nicméně navzdory odlišnému iniciálnímu patogenetickému mechanismu jsou typy IPP v rámci SNP podobné radiologicky i patologicky idiopatickým intersticiálním pneumoniím, případně vaskulitickému postižení (tab. 2) a mají obdobnou odpověď na protizánětlivou či antifibrotickou léčbu s ohledem na typ postižení.1
Vyšetření
Klinický obraz
Klinicky se většina IPP u SNP projevuje progredující námahovou a posléze klidovou dušností, snadnou unavitelností, kašlem a v pozdějších fázích při nastupující hypoxemii i cyanózou. U některých IPP (fenotyp plicní fibrózy obvyklého typu – UIP) se také vyskytují fenotypové projevy, jako jsou paličkovité prsty s nehty tvaru hodinového sklíčka a poslechový fenomén krepitu slyšitelný nad plicními bazemi. Hemoptýza obvykle nebývá v popředí symptomů plicního postižení v rámci SNP, může se zřídka vyskytovat v rámci kapilaritidy u SLE, kde pak může být dominujícím projevem.1,2
Laboratorní nálezy
Laboratorní nálezy u IPP při SNP většinou nejsou patognomické a ve většině případů nenapomohou diagnóze. Výjimkou jsou některé autoprotilátky, které predikují vyšší pravděpodobnost IPP u SNP (protilátky proti topoizomeráze u SSc). V případě IPP spojených s alveolární hemoragií (kapilaritida u SLE) bývá přítomna anémie, většinou sideropenická. Naopak v případě pokročilých IPP s hypoxemií je často přítomna nápadná polyglobulie.1
Funkční vyšetření plic
IPP u SNP jsou většinou charakterizovány restriktivní ventilační poruchou, snížením hodnot transfer faktoru (TLCO) a sníženou plicní poddajností. Normální plicní funkce však nevylučují IPP v rámci SNP. Spiroergometrie může odhalit jak iniciální plicní postižení, tak i plicní hypertenzi v rámci SNP. Vyšetření saturace periferní krve kyslíkem a krevní plyny diagnostikují hypoxemii a jsou odrazovým plicním můstkem pro indikaci domácí oxygenoterapie, je‑li indikována.1
Zobrazovací metody IPP u SNP
Základním radiologickým vyšetřením IPP v rámci SNP je výpočetní tomografie hrudníku s vysokou rozlišovací schopností (HRCT). Plicní postižení u SNP kopíruje jednotlivé podtypy idiopatických intersticiálních pneumonií, někdy bývají přítomny i známky postižení pleury. Obvykle se u SNP vyskytují fenotypy UIP, nespecifická intersticiální pneumonie (NSIP), lymfocytární intersticiální pneumonie (LIP) a organizující se pneumonie (OP) (tab. 2). Méně často se vyskytuje obraz difuzní alveolární hemoragie (DAH) nebo granulomatózního plicního postižení (tab. 3). Specifickým podtypem IPP je progredující plicní fibróza (PPF), kterou určuje kromě radiologického a histologického obrazu i progrese rozsahu fibrózních změn a zhoršování plicní funkce a symptomů v čase.1,2,4

Bronchoskopie a bronchoalveolární laváž (BAL)
U řady jedinců s IPP není bronchoskopie ani BAL prováděna, neboť nejsou nutné pro diagnózu ani management onemocnění. Pokud však potřebujeme odlišit infekční postižení či nádorový proces, pak je jednoznačně bronchoskopie s BAL a cílenými odběry indikována, Nález v tekutině získané BAL (BALTe) je variabilní a záleží na patologicko‑radiologickém fenotypu IPP. Zvláštností je, že i fenotyp fibrotický v rámci SNP mívá v BALTe větší zastoupení lymfocytů než obdobný fenotyp v rámci IPF.1
Plicní biopsie u IPP u SNP
Ve většině případů IPP u SNP není chirurgická plicní biopsie nutná, pokud je fenotyp postižení dobře definován dle klinického obrazu, funkčního vyšetření, HRCT, případně BAL. Pokud však je typ IPP nejistý a na jeho přesné definici závisí další léčba (kupříkladu IPP v rámci SNP versus difuzní nádor či infekce), je kryobiopsie (bronchoskopická biopsie, kdy tkáň získáme přimražením na kryosondu zavedenou do hloubky plicního parenchymu – obr. 1), případně chirurgická plicní biopsie nezbytná (samozřejmé je však zvážení celkového zdravotního stavu pacienta a pokročilosti IPP).1

Histopatologie
Histopatologický obraz SNP v plicním parenchymu zahrnuje postižení dýchacích cest, alveolů, plicních cév, pleury a hrudní stěny. Intersticiální plicní postižení je nejčastěji reprezentováno obrazem různých forem NSIP, méně často UIP. Při diagnostice plicního postižení v rámci SNP je nejdůležitější úzká klinicko‑patologická korelace.
Screening IPP v rámci SNP
V případě diagnostiky IPP u pacientů se SNP jsou v podstatě dva scénáře: 1. screening IPP u stran respiračního traktu asymptomatického pacienta se SNP nebo 2. diagnostická sestava vyšetření u pacienta se SNP s respiračními symptomy. Screening provádíme v těch případech, kdy je vysoká pravděpodobnost IPP vzhledem k základní diagnóze. Optimálním screeningovým nástrojem je HRCT hrudníku, nicméně dosud panuje debata, zda je adekvátním a bezpečným nástrojem u mladých pacientů s ohledem na radiační zátěž, pokud se vyšetření periodicky opakuje. Nedílnou součástí screeningu a diagnostiky nitrohrudního postižení u SNP je i screening a diagnostika plicní hypertenze, zejména u chorob s vysokým rizikem, jako je SSc.1,2,4
Léčba IPP v rámci SNP a sledování
Rozhodnutí o zahájení a typu léčby IPP při SNP záleží pak na řadě faktorů. Rozhodující je zejména typ plicního postižení, zda jde o zánětlivý, kombinovaný, nebo fibrotický proces, zejména charakteru PPF. Dle toho pak volíme léčbu protizánětlivými léky a imunosupresivy (kortikosteroidy, azathioprin, mykofenolát mofetil), případně biologickou léčbu – rituximab – anebo léčbu antifibrotickou – nintedanib. V případě rozsáhlého progredujícího fibrotického postižení plic je nutné včas zvážit případně transplantaci plic (tab. 3). Pacienti s IPP‑SNP by měli být sledováni a léčeni v centrech pro diagnostiku a léčbu IPP ve spolupráci s revmatologem a případně nefrologem, kardiologem či neurologem při dalších orgánových postiženích. V terminálních stadiích IPP pak indikujeme obvykle dlouhodobou domácí oxygenoterapii (DDOT) a paliativní péči.5–8
Prognóza
Prognóza jednotlivých podtypů plicních postižení u SNP se značně liší a je závislá zejména na typu IPP (zánětlivý versus fibrotický), funkčním postižení a také na komplikujících faktorech (postižení cév, plicní infekce). Nejzávažnější je fenotyp UIP a obecně PPF, i když v rámci SNP může mít lepší prognózu než idiopatická plicní fibróza a idiopatické PPF. I v případě IPP v rámci SNP může dojít k tzv. akutním exacerbacím, které bohužel ve většině případů navzdory komplexní léčbě končí fatálně.1,2,4
Plicní postižení při systémové sklerodermii
Systémová sklerodermie (SSc) je komplexní systémová nemoc s extenzivní fibrózou, vaskulárními změnami a autoprotilátkami proti různým buněčným antigenům – antitopoizomeráza I (Scl‑70), anticentromerové, antinuklární autoprotilátky, autoprotilátky proti receptoru pro destičkový růstový faktor (PDGFR). Plicní postižení je u SSc velmi časté, dochází k němu až v 70 % případů, proto je nutné provádět screening plicního postižení metodou HRCT. Vyskytuje se hlavně u pacientů s pozitivními autoprotilátkami proti topoizomeráze (ATA), anticentromerové autoprotilátky (ACA) jsou spojeny hlavně s plicním cévním postižením.
Nejčastějším typem plicního postižení u SSc je NSIP, méně pacientů má obraz UIP, někdy se objevuje obraz tzv. centrilobulární fibrózy, která může mít souvislost s opakovanými aspiracemi při gastroezofageálním refluxu. Méně častými fenotypy postižení jsou OP, LIP, pleuropulmonální fibroelastóza (PPFE) nebo neklasifikovatelná plicní fibróza. Nutné je myslet i na případnou plicní arteriální hypertenzi (PAH) u SSc, která modifikuje obraz plicního postižení nebo se vyskytuje izolovaně a významně zhoršuje prognózu pacientů. Častější je u nemocných se syndromem CREST (C – calcinosis cutis – podkožní kalcifikace, R – Raynaudův fenomén, E – porucha motility ezofagu, S – sklerodaktylie, T – teleangiektazie).
Pro praktické účely léčby a predikce prognózy pro samotný IPP u SSc je pak užitečné dělení na zánětlivé (nefibrotické) postižení a fibrotické postižení, zejména charakteru PPF. Při zánětlivém postižení je doporučován mykofenolát mofetil (MMF), případně cyklofosfamid, obvykle v pulzním režimu 500–1 000 mg/m2/měsíc. Při progredujícím fibrotickém postižení je indikována léčba nintedanibem s tím, že může být ponechán MMF, pokud je vhodné mít kombinaci s protizánětlivou léčbou. U progredujících zánětlivých forem refrakterních na výše uvedené typy léčby je možné indikovat rituximab. Pirfenidon nemá dostatek důkazů pro použití u PPF SSc, nedá se tedy zatím klinicky využít (obr. 2).5–7,9

Prognóza plicního SSc IPP je významně lepší než u IPF, deset let přežívá 70 % pacientů. Pokud je však hodnota TLCO snížena pod 40 %, pak pětileté přežití klesá pod 10 %. Přežití u obou základních fenotypů plicního parenchymatózního postižení (UIP a NSIP) v rámci SSc je na rozdíl od idiopatických bez významného rozdílu. Při plicním postižení typu UIP pět let přežívá 82 % a při NSIP pět let přežívá 91 % pacientů. Špatným prognostickým faktorem je pokles hodnoty TLCO v posledních třech letech a hypereozinofilie v BALTe. Dalším významným faktorem zhoršujícím přežití je plicní hypertenze, zvláště je‑li nepoznaná, a tudíž neléčená.
Postižení plic při revmatoidní artritidě
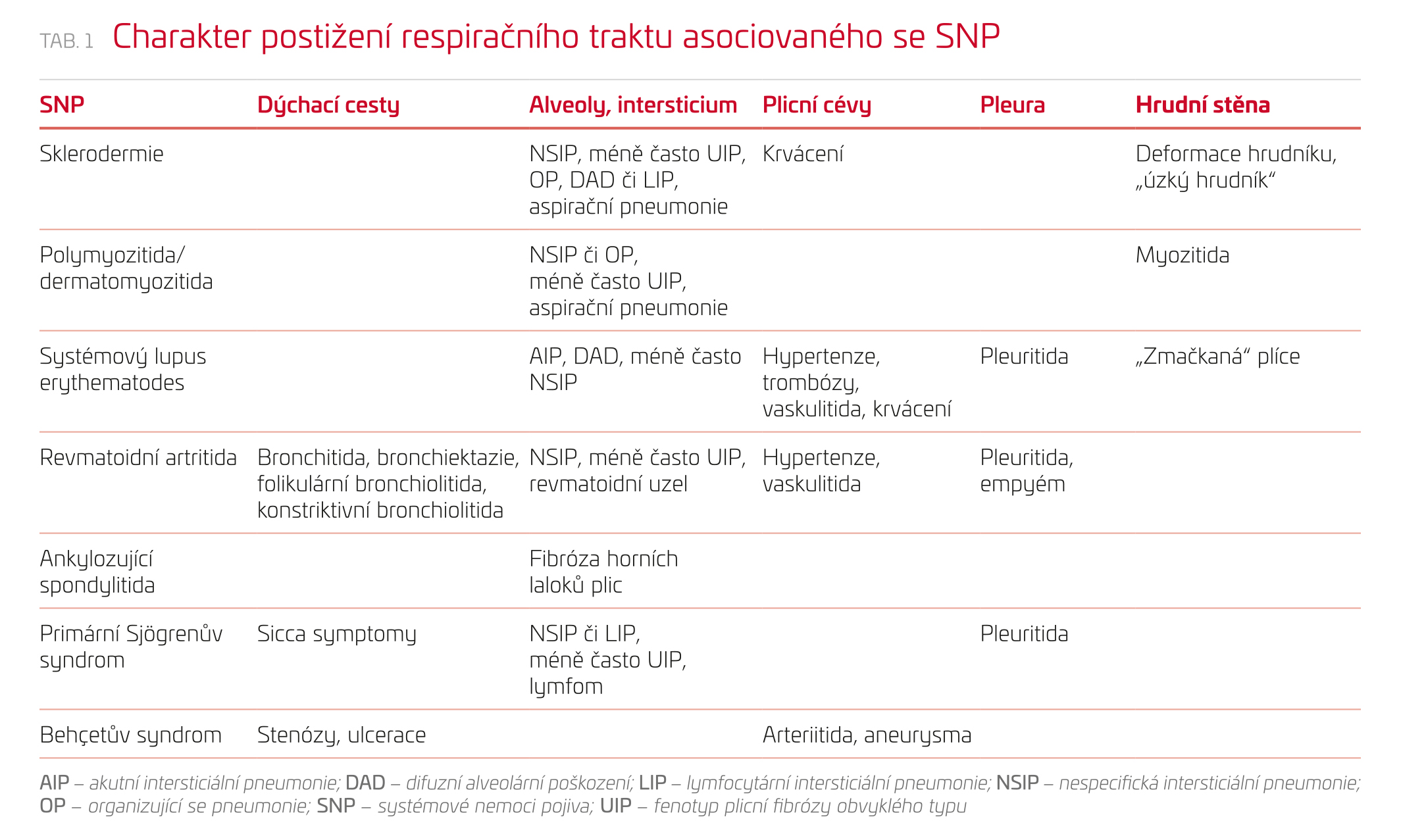
Revmatoidní artritida je relativně častým onemocněním, které postihuje přibližně 1 % populace. Plicní postižení u RA je relativně časté a náchylnost k němu je geneticky svázána s HLA genotypem HLA‑B8 a HLA‑Dw3. Fibrotický typ postižení u RA se vyskytuje častěji u pacientů s vysokým titrem revmatoidního faktoru (RF) a s revmatickými uzly v plicích. V rámci RA na rozdíl od ostatních SNP většinou zachytíme postižení plic typu UIP (56 %), méně často smíšenou nebo fibrotickou NSIP (33 %) (obr. 3). Někdy může být RA provázena obliterující bronchiolitidou (OB) a folikulární bronchitidou (FB) s OP nebo bez OP (11 %). Zřídka vídáme LIP a také nekavitující či kavitující revmatické uzly. Někdy může onemocnění akutně exacerbovat pod obrazem difuzního alveolárního poškození (DAD). Z dalších postižení respiračního systému můžeme pozorovat postižení pleury, někdy až s obrazem empyému rezistentního na léčbu, krikoarytenoidní artritidu, bronchiektazie, Caplanův syndrom, lymfoidní hyperplazii a vaskulitidu plicních cév.
U většiny pacientů s RA se plicní postižení manifestuje ve věku mezi 50 a 60 lety. Také podobně jako u jiných SNP se plicní postižení může vyskytnout i izolovaně, bez klinických projevů kloubního postižení. U mužů a kuřáků s RA se nejčastěji setkáme s fenotypem UIP, ženy mají častěji obraz NSIP.
Vzhledem k závažnosti a četnosti plicního postižení při RA je nutné jeho aktivní vyhledávání v populaci pacientů s RA pomocí klinického, funkčního a radiologického vyšetření (HRCT). Diagnóza je postavena obvykle na diagnóze RA, typickém radiologickém obraze a funkčních změnách. BAL a plicní biopsie nejsou většinou indikovány. V rámci RA kromě různých fenotypů plicní manifestace můžeme pozorovat i infekční komplikace v rámci imunosupresivní léčby a polékové postižení plic, především metotrexátovou pneumonitidu, která se vyskytuje u 0,3–11,6 % nemocných s RA léčených MTX.
Základem léčby RA IPP jsou imunosupresiva. Důležité je rozhodnutí, zda léčit, tedy zda nežádoucí účinky léčby nepřevýší její potenciální profit, ať už ve smyslu rizika progrese nemoci při plicní toxicitě léku, nebo ve smyslu infekčních komplikací. Základními imunosupresivy v případě IPP v rámci RA jsou mykofenolát mofetil, azathioprin, cyklofosfamid a případně rituximab (tab. 3). U minimálního postižení plic lze zvážit i metotrexát, případně leflunomid. Plicní toxicita, ze které byl zejména metotrexát dlouhodobě obviňován, nebyla klinickými pozorováními v závažném rozsahu potvrzena. Kortikosteroidy, které byly doposud zvažovány jako metoda první volby u IPP v rámci RA, vykazují vyšší riziko úmrtí ze všech příčin ve srovnání s azathioprinem a zejména s mykofenolát mofetilem. Jejich použití by mělo být tedy velmi bedlivě zvažováno. Anti‑TNF alfa léčba (infliximab, adalimumab) může mít na plicní parenchym u RA dvojí efekt, může sice potlačovat IPP, nicméně byly popsány i případy, kdy tato léčba mohla být příčinou zhoršení RA IPP. Nové metody biologické léčby RA mohou pravděpodobně přinášet i pozitivní efekt na IPP RA, zejména léčba ovlivňující signalizaci interleukinu 6, kupříkladu tocilizumab, nicméně není zatím dostatek důkazů pro užití tocilizumabu pro léčbu RA IPP, a neměl by tedy být v této indikaci primárně podáván. Pro pacienty s PPF v rámci RA je vhodné zvážit antifibrotickou léčbu nintedanibem.5,6,8–10
Prognóza závisí na fenotypu plicního postižení, významně horší je u těch, kteří mají fenotyp UIP, oproti pacientům s NSIP. Při porovnání prognózy pacientů s IPF a s plicním postižením při RA typu UIP nebyl pozorován významný rozdíl v délce přežívání.
Postižení plic při IIM
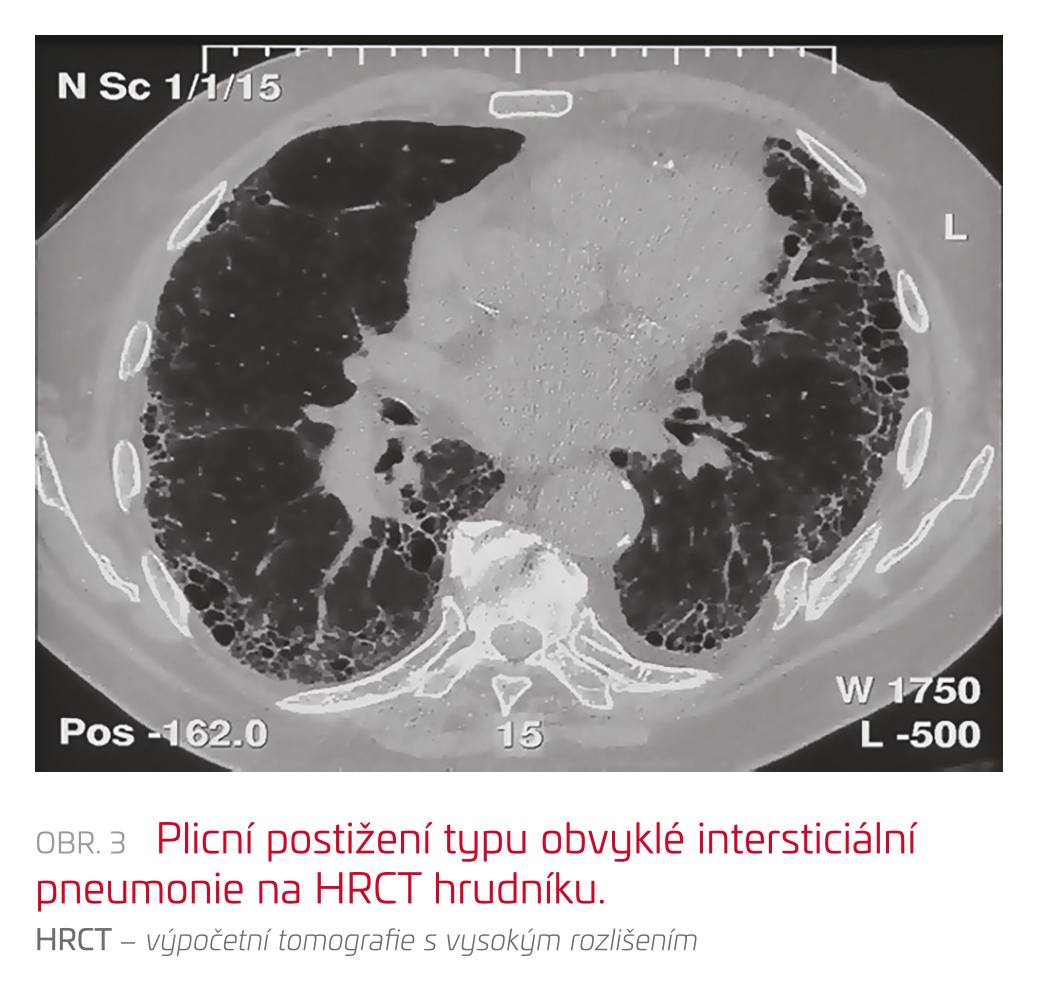
Idiopatické zánětlivé myopatie/myozitidy (IIM), dříve označované jako komplex polymyozitidy/dermatomyozitidy (PM/DM), se vyskytují relativně často s plicním postižením, většinou v rámci tzv. antisyntetázového syndromu. Ten je charakterizován přítomností protilátek proti aminoacyl‑tRNA syntetázám, nejčastější je anti‑histidyl (Jo‑1), a některým z následujících klinických symptomů – myozitidou, IPP, artritidou nebo artralgiemi, Raynaudovým fenoménem, „rukama mechanika“ (obr. 4) a horečkou. Někdy může být přítomna i porucha motility jícnu. Někdy můžeme detegovat i autoprotilátky proti jiným tRNA syntetázám, anti‑threonyl (PL‑7), anti‑alanyl (PL‑12) a jiným, proti kterým se však rutinně obvykle netestuje.
Plicní postižení se vyskytuje u myozitid ve 20–40 % případů a až ve třetině případů může předcházet klinické akutní intersticiální pneumonii (AIP). Obraz může být modifikován svalovým postižením dechových svalů a svalů hrtanu (opakované aspirace). V případě akutního průběhu je většina pacientů přijímána k hospitalizaci pro dušnost, naopak chronické IPP v rámci myozitid mohou probíhat i klinicky němě. Většinou je přítomno postižení kůže a svalů, někdy se však můžeme setkat s amyopatickou formou, někdy může chybět i kožní postižení. V laboratorních nálezech bývá zjišťována zvýšená hodnota myoglobinu, kreatinfosfokinázy a jsou přítomny autoprotilátky, nejčastěji anti‑Jo‑1. V radiologickém nálezu najdeme při akutním průběhu s dušností obvykle obraz AIP, který je klinicky spojen až se syndromem akutní dechové tísně (ARDS), při chronických průbězích bývá častější OP či NSIP. V BALTe bývá zvýšený celkový počet buněk a v rozpočtu je zvýšené zastoupení lymfocytů, a to hlavně u akutních a subakutních forem. Diagnóza je obvykle stanovena na základě klinicko‑radiologického nálezu, důležité je myslet na IIM IPP v případě akutní pneumonie neustupující po antibiotikách, případně antivirotikách, zvláště jsou‑li přítomny kožní příznaky a laboratorní vyšetření dokládá zvýšené hodnoty svalových enzymů a průkaz protilátek proti aminoacyl‑tRNA syntetázám.
Léčebně podáváme kortikoidy v dávce 0,75–1 mg/kg/den, u akutních forem volíme pulzní podání v dávce 1 000 mg metylprednisolonu denně v úvodu léčby, zásadní je přidat i imunosupresiva, jmenovitě cyklofosfamid v pulzním režimu, alternativou může být azathioprin nebo mykofenolát mofetil. Dávky systémových kortikosteroidů by měly být co nejdříve snižovány vzhledem k riziku nežádoucích účinků při dlouhodobém užívání. U akutních fulminantních průběhů můžeme zkusit intravenózní podání gamaglobulinů. Při formách rezistentních na uvedenou léčbu je možné podat rituximab, případně anti‑JAK inhibitor tofacitinib.5,6,8,9,11
Akutní IPP u myozitid může mít i fulminantní průběh navzdory léčbě, až polovina těchto pacientů zemře na respirační selhání do 1–2 měsíců (pětileté přežití 35 %). V případě amyopatických forem může být plicní postižení ještě závažnější s mortalitou 75–86 %. Chronické formy IPP mají prognózu jednoznačně lepší, pět let přežívá 100 % pacientů. Vzhledem k závažnosti akutního plicního postižení při myozitidách musíme po IPP aktivním screeningem pátrat a musíme i myslet na možný IIM IPP u klinicky nemanifestní myozitidy při závažných difuzních pneumoniích nereagujících na antibiotika.3
Plicní postižení při systémovém lupus erythematodes
Systémový lupus erythematodes (SLE) je systémovou autoimunitní nemocí s multisystémovým postižením s četnými sérologickými abnormalitami. Patogeneticky bývá charakterizován jak zánětlivou, tak fibrotickou složkou a výsledný obraz závisí na kombinaci těchto dvou fenotypů IPP. V rámci postižení respiračního traktu mohou být postiženy jak dýchací cesty, tak plicní parenchym, vaskulatura, pleura i hrudní stěna. Nicméně u SLE je plicní postižení málo časté. Je popsána vazba plicního postižení na pozitivitu anti‑Ro/SSa. Nejčastějším typem postižení je NSIP, akutní lupoidní pneumonitida a alveolární hemoragie, vzácně se může vyskytnout i fibróza.
IPP u SLE nejčastěji mívá podobu NSIP, méně často OP či difuzní amyloidózy, vzácně UIP a LIP. Klinicky se projeví námahovou dušností a kašlem. Poslechově může být slyšitelný krepitus. Vzácnou plicní komplikací je i AIP s klinickým obrazem ARDS, který se objevuje u 5–15 % pacientů se SLE. Mezi jeho vyvolávající faktory patří infekce a antifosfolipidový syndrom. Velmi závažnou plicní manifestací SLE je difuzní alveolární hemoragie (DAH), která se vyskytuje u 1–5 % nemocných se SLE. Náhlý nárůst dušnosti s novým vznikem opacit mléčného skla na HRCT hrudníku je vždy podezřelý z DAH, zvláště je‑li pozorován pokles hodnoty hematokritu. Někdy je DAH spojena i s nefritidou. Postižen může být i plicní cévní oběh, což se manifestuje nejčastěji obrazem plicní hypertenze, a to u 6–14 % pacientů se SLE. Často se pak u těchto pacientů objevuje i Raynaudův fenomén, který poukazuje na vaskulitidu jako příčinu PAH i postižení periferní cirkulace. S postižením plicních cév souvisí i syndrom přechodné hypoxemie, způsobený pravděpodobně agregací neutrofilů indukovanou aktivací komplementu v kapilárách. Zřídka se můžeme setkat s obrazem plicní venookluzivní nemoci (PVOD). V rámci extraparenchymatózního postižení respiračního traktu je nejčastěji postižena pleura. Mezi klinické projevy jejího postižení patří bolest na hrudi vázaná na dechové pohyby a/nebo narůstající dušnost při tvorbě výpotku. Postižení pleury bývá až u třetiny pacientů se SLE, nemusí být vždy klinicky manifestní a může být nalezeno až při pitvě. Vzácně bývají postiženy dýchací svaly a bránice. Syndrom postižení bránice byl nazván „shrinking lung syndrome“ – SLS čili syndrom srážejících se plic. Jeho podkladem je fibrotizace bránice a její atrofie. Dušnost u SLE může být způsobena i trombotickými a tromboembolickými komplikacemi na podkladě defektů plicních cév způsobených antifosfolipidovými protilátkami – lupus antikoagulans (LA) a antikardiolipinový syndrom i autoprotilátky. Netrombotickými projevy antifosfolipidového syndromu mohou být PAH, DAH, ARDS nebo léze srdečních chlopní.
Pro diagnózu a diferenciální diagnózu plicního postižení u SLE je důležitá kromě klinického vyšetření zejména HRCT hrudníku. Z IPP nejčastěji vidíme NSIP s obrazem opacity mléčného skla s retikulací, s drobnými cystami a uzly. Akutní lupusová pneumonitida se manifestuje ložisky opacit mléčného skla v plicní periferii a v plicních bazích. Bronchoalveolární laváž je významně nápomocna pro odlišení DAH od IPP postihujících primárně plicní parenchym a k odlišení infekčních komplikací.
Diagnóza je postavena na základě anamnézy SLE s některým z výše popsaných radiologických fenotypů plicního postižení s typickým funkčním postižením. BALTe nám pomůže odlišit hlavně DAH a infekci. Důležité je odlišit i polékové postižení plic. Chirurgická plicní biopsie je indikována zřídka.
Stran terapie bohužel není dostatek údajů o léčbě IPP u SLE, které by byly získány na základě randomizovaných klinických studií. Opíráme se tedy většinou o zkušenosti z léčby IPP u SSc a o klinickou zkušenost. Základem léčby IPP při SLE v první linii by měl být dle Evropské ligy proti revmatismu (EULAR) hydroxychlorochin, obvykle jsou přidávány kortikosteroidy. Případně můžeme kombinovat kortikosteroidy s imunosupresivy (cyklofosfamid v úvodu s přechodem na mykofenolát mofetil nebo azathioprin v udržovací fázi). V případě refrakterní nemoci nebo LIP je vhodné zvážit rituximab. Při DAH je nutné zahájit léčbu vysokými dávkami intravenózních (i.v.) kortikoidů (1 g metylprednisolonu tři dny po sobě) s následným podáváním 60 mg prednisonu denně. Současně je podáván cyklofosfamid 500–1 000 mg/m2 i.v. každé 4 týdny. Někdy pro refrakterní DAH indikujeme plazmaferézu. Zvýšenou hodnotu alkalické fosfatázy léčíme také jako DAD, případně léčíme interkurentní infekci, pokud je prokázána. Prvním lékem registrovaným k terapii SLE je belimumab, rekombinantní monoklonální protilátka proti faktoru aktivujícímu B lymfocyty, nicméně o efektu tohoto přípravku v prevenci a léčbě IPP u SLE není dostatek informací. V případě PPF u SLE je indikována antifibrotická léčba nintedanibem. PAH u SLE v řadě případů odpovídá na imunosupresivní léčbu, pokud nikoli, je namístě specifická léčba v centru pro plicní hypertenzi.5,6,8,9,12
Desetileté přežití je u SLE IPP udáváno kolem 90 %. Příčinou úmrtí jsou nejčastěji aktivita SLE, trombotické komplikace a infekce. Velmi špatná je prognóza DAH, 50 % nemocných umírá již za hospitalizace pro tuto příhodu.
Plicní postižení u Sjögrenova syndromu
Sjögrenův syndrom (SS) je chronickým zánětlivým autoimunitním onemocněním s lymfocytární infiltrací exokrinních žláz, s následným sicca syndromem. Může se objevit buď izolovaně, nebo ve spojení s jiným autoimunitním onemocněním. V průběhu SS se může objevit postižení řady orgánů – plic, ledvin a malých cév.
Plicní postižení může probíhat pod různými fenotypy s různou prognózou, vyskytuje se u 9–75 % pacientů, v závislosti na použité metodě detekce IPP. Nejčastějším fenotypem postižení u SS je NSIP, celulární podtyp, zřídka vídáme LIP a můžeme se setkat i s maligní lymfoproliferací. Někdy se setkáme s fenotypem UIP a vzácně i s plicní amyloidózou. Někdy mohou být postiženy jen bronchioly s obrazem difuzní bronchiolitidy, někdy je přítomen obraz OP.
Věk pacientů v době diagnózy je obvykle kolem 60 let, většinou se jedná o ženy. Pacienti si obvykle stěžují na potíže vyplývající ze sicca syndromu. Postižení plic se pak projevuje kašlem a dušností, někdy bývají přítomny i systémové příznaky (bolesti kloubů a horečky). Poslechově bývá slyšitelný krepitus u 64 % pacientů. Typickou autoprotilátkou pro SS je ANA a anti‑SS‑A/Ro. U řady pacientů pozorujeme také polyklonální hypergamaglobulinemii, zvýšenou sedimentaci a zvýšené titry RF. U poloviny pacientů se vyskytuje i pozitivita anti‑SS‑B/La.
Častým radiologickým nálezem u SS IPP je obraz odpovídající NSIP, vzácnější je UIP a OP, ojediněle je spojen s obrazem LIP. V popředí změn je opacita mléčného skla, nepravidelná retikulace a ložiska kondenzace, vzácnější jsou uzlovité léze a ztluštění interlobulárních sept. Může být přítomna i voštinovitá plíce a tenkostěnné cysty. Příčina cyst není jasná, jde zřejmě o ventilový mechanismus podmíněný buněčnou infiltrací v oblasti bronchiolárních struktur. U části nemocných se zjišťuje v exspiraci „air‑trapping“, odpovídající obliterující bronchiolitidě. V BALTe je u většiny pacientů zvýšeno zastoupení lymfocytů, ale i neutrofilů a eozinofilů.
Diagnóza SS IPP je stanovena na základě diagnózy SS, radiologického a funkčního postižení typického pro IPP, podpořeného BAL. Pokud máme podezření na LIP nebo lymfoproliferaci, měla by být provedena chirurgická biopsie.
Pro léčbu IPP u SS existují pouze klinické zkušenosti, na jejichž základě doporučujeme léčbu. Na rozdíl od IPP u SSc je na prvním místě doporučována léčba systémovými kortikosteroidy, avšak s brzkým snižováním dávky a s přidáním kortikosteroidy šetřících imunosupresiv, jako je mykofenolát mofetil nebo azathioprin, případně cyklofosfamid. Plicní lymfom je samozřejmě léčen hematoonkology specifickými režimy. Pětileté přežití pacientů se SS s plicním postižením dosahuje 85 %, špatnou prognózu mají pacienti s maligním lymfomem nebo s rozsáhlým fibrotickým postižením plic. Pro pacienty s PPF v rámci SS je nově doporučena, stejně jako u ostatních progredujících fibróz, antifibrotická léčba nintedanibem, která by mohla zlepšit životní výhled i u těchto jedinců.5,6,8,9,13
Nediferencované onemocnění pojiva (UCTD) a IPP
Nediferencované onemocnění pojiva (undifferentiated connective tissue disease, UCTD) je popsáno jako soubor symptomů nasvědčujících systémové nemoci pojiva, které však nelze přesně zařadit do žádné ze SNP. Velmi často je součástí UCTD plicní postižení s obrazem NSIP. Dokonce se spekuluje o tom, že řada tzv. idiopatických NSIP je vlastně součástí UCTD. Fenotypově se mimo jiné liší od idiopatických NSIP tím, že mají lepší prognózu. Prakticky 50 % pacientů s NSIP splňuje tato kritéria oproti pouze 5 % pacientů s definitivní diagnózou IPF.
V BALTe najdeme výraznou lymfocytózu, více prominentní než u idiopatické NSIP. Pacienti s NSIP v rámci UCTD mají významně lepší prognózu než pacienti s iNSIP. V roce 2015 byla navržena nová klinická jednotka, a to intersticiální pneumonie s autoimunitními rysy (IPAF), která má obdobné charakteristiky jako UCTD. De facto by IPAF anebo i UCTD mohlo odpovídat SNP s ne zcela vyjádřeným systémovým postižením a s dominujícím postižením plic.5,6,8,9
Závěr
U pacientů se systémovou nemocí pojiva je třeba vždy myslet na intersticiální plicní postižení, neboť jde o velmi častý projev těchto nemocí, a navíc významně ovlivňuje prognózu a životní výhled pacienta. Základním diagnostickým a screeningovým nástrojem je HRCT hrudníku, určení stupně funkčního postižení a jeho monitoraci v čase slouží spirometrie a plicní difuze. Léčba závisí na charakteru IPP, může být buď protizánětlivá, nebo antifibrotická. V pokročilých stadiích je pak u některých nemocných indikována transplantace plic.
Literatura
- Koziar Vašáková M, Polák J, Kočová E, Matěj R. Intersticiální plicní procesy. 3. aktualizované a rozšířené vydání. Maxdorf: Praha, 2024.
- Fischer A, du Bois R. Interstitial lung disease in connective tissue disorders. Lancet 2012;380:689–698.
- Johnson SR, Bernstein EJ, Bolster MB, et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guideline for the screening and monitoring of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthritis Care Res 2024;76:1070–1082.
- Jeganathan N, Sathananthan M. Connective tissue disease‑related interstitial lung disease: prevalence, patterns, predictors, prognosis, and treatment. Lung 2020;198:735–759.
- Johnson SR, Bernstein EJ, Bolster MB, et al. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) guideline for the treatment of interstitial lung disease in people with systemic autoimmune rheumatic diseases. Arthritis Care Res 2024;76:1051–1069.
- Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019;381:1718–1727.
- Raghu G, Montesi SB, Silver RM, et al. Treatment of Systemic Sclerosis‑associated Interstitial Lung Disease: Evidence‑based Recommendations. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med 2024;209:137–152.
- Ahmed S, Handa R. Management of Connective Tissue Disease‑related Interstitial Lung Disease. Curr Pulmonol Rep 2022;11:86–98.
- Barnes H, Holland AE, Westall GP, et al. Cyclophosphamide for connective tissue disease‑associated interstitial lung disease. Cochrane Database Syst Rev 2018;1:CD010908.
- Cassone G, Manfredi A, Vacchi C, et al. Treatment of Rheumatoid Arthritis‑Associated Interstitial Lung Disease: Lights and Shadows. J Clin Med 2020;9:1082.
- Hallowell RW, Paik JJ. Myositis‑associated interstitial lung disease: a comprehensive approach to diagnosis and management. Clin Exp Rheumatol 2022;40:373–383.
- Richter P, Cardoneanu A, Dima N, et al. Interstitial Lung Disease in Systemic Lupus Erythematosus and Systemic Sclerosis: How Can We Manage the Challenge? Int J Mol Sci 2023;24:9388.
- Luppi F, Sebastiani M, Silva M, et al. Interstitial lung disease in Sjögren’s syndrome: a clinical review. Clin Exp Rheumatol 2020;38 (Suppl 126):291–300
- Kategorie: Názor hosta
- Klíčová slova: antifibrotická léčba; plicní postižení; protizánětlivá léčba; systémové nemoci pojiva