aHUS a C3 nefropatie: doporučení pro diagnostiku a léčbu
Souhrn
Diagnostika a léčba onemocnění spojených s nadměrnou aktivací komplementu se stále posouvá dopředu, nicméně s ohledem na to, že jde o vzácná onemocnění, je povědomí o těchto chorobách poměrně malé. Poškození ledvin mohou tyto choroby způsobovat buď formou trombotické mikroangiopatie (všechny formy hemolyticko‑uremického syndromu), nebo depozicí složek komplementu (zejména C3) v různých částech ledvin (C3 glomerulopatie). Pochopení základních patogenetických mechanismů nám může pomoci v diagnostice, ale i v léčbě těchto stavů.
V roce 2015 se sešel panel expertů zastoupených v iniciativě KDIGO (Kidney Disease: Improving Global Outcomes) na konferenci věnované sporným otázkám, kde se snažili definovat problémy spojené s onemocněními komplementu (zejména atypického hemolyticko‑uremického syndromu – aHUS – a C3 glomerulopatie – C3G) a navrhnout doporučení pro jejich diagnostiku a léčbu. Závěry této konference pak publikovali v roce 2017 v souhrnném článku.1
Jak u aHUS, tak u C3G hraje aktivace komplementu zásadní roli v patogenezi onemocnění. Atypický hemolyticko‑uremický syndrom je velmi vzácné onemocnění s incidencí přibližně 0,5 pacientů/milion, které se většinou manifestuje akutním poškozením ledvin, trombocytopenií, trombotickou mikroangiopatií (TMA) s hemolytickou anémií. Ostatní onemocnění patřící do skupiny TMA zahrnují hemolyticko‑uremický syndrom způsobený infekcí Escherichia coli produkující shigatoxin (STEC‑HUS), trombotickou trombocytopenickou purpuru (TTP), antifosfolipidový syndrom (APS) a řadu dalších, většinou systémových onemocnění. Více než 50 % nemocných s aHUS má vrozenou či získanou abnormalitu v komplementu, která způsobuje aktivaci alternativní cesty komplementu a aktivaci endotelu. Existují ale i vrozené abnormality proteinů nepatřících do komplementové kaskády, např. mutace v genu DGKE (diacylglycerol kinase ξ), které mají fenotyp shodný s aHUS. Do nedávné doby byla prognóza nemocných s aHUS velmi špatná a u řady nemocných onemocnění progredovalo do terminálního selhání ledvin během dvou let od stanovení diagnózy. Se zavedením inhibitorů komplementu do léčby se významně zlepšila možnost kontrolovat průběh onemocnění a zabránit rozvoji ireverzibilního renálního poškození.
C3 glomerulopatie patří rovněž mezi extrémně vzácná onemocnění (incidence přibližně 1,0 pacient/milion/rok), pro které je typické progredující renální postižení spojené s nekontrolovanou aktivací komplementové kaskády s depozicí C3 v glomerulech. Geneticky navozený či získaný defekt postihuje zejména C3 konvertázu a je rovněž spojen s aktivací alternativní cesty komplementu. V závislosti na interindividuální variabilitě pak vznikají dvě různé podskupiny tohoto onemocnění: nemoc s ukládáním denzních depozit (DDD) a C3 glomerulonefritida (C3GN). Rozlišení obou forem onemocnění je možné jen pomocí renální biopsie.
Renální patologie
Atypický hemolyticko‑uremický syndrom
Atypický hemolyticko‑uremický syndrom je typickým představitelem TMA a může způsobovat jak akutní, tak chronické poškození ledvin. Poškozené bývají zejména glomeruly, arterioly i arterie, druhotně pak dochází k poškození i dalších struktur ledvin, např. tubulů a intersticia. Mezi akutní formy poškození v renální biopsii (RB) patří zejména všudypřítomné tromby, intramurální depozita fibrinu, edém a „odloupání“ buněk endotelu, mezangiolýza, přítomnost fragmentovaných erytrocytů či vznik mikroaneuryzmat. Mezi chronické formy poškození lze zařadit rozšíření stěny kapilárních kliček až se zdvojením jejich kontury, rozšíření subendoteliálních prostor, hyalinní depozita v arteriolách či fibrózní ztluštění intimy cév s jejich koncentricky laminární strukturou (vzhled cibulových vrstev jako projev myofibrilární proliferace). K diagnóze přispívá přítomnost depozit pozitivně se barvících na C5b‑9, jež se ale běžně nestanovuje. Přítomnost známek TMA v renální biopsii významným způsobem přispívá k diagnostice těchto chorob, ale jednotlivé formy TMA nemají své specifické rysy, a tudíž z RB nelze určit etiologii vzniklé TMA. Renální biopsie ale může pomoci v odhadu rozsahu poškození ledvin a předpokládané renální prognózy.
C3 glomerulopatie
Pro C3G je typické ukládání fragmentů C3 složky komplementu ve formě depozit v ledvinách při imunofluorescenčním vyšetření (IF), zatímco ostatní vyšetřované složky jsou negativní či silně potlačeny (zejména imunoglobuliny či C1q složka komplementu). Imunofluorescenční vyšetření je klíčové pro stanovení diagnózy C3G. Podle místa, kde se depozita dominantně kumulují při vyšetření elektronovou mikroskopií, pak mluvíme o DDD (denzní depozita intramembranózně v bazální membráně glomerulů) anebo o C3GN (světle denzní amorfní depozita zejména v mezangiu, paramezangiálně, subendoteliálně a subepiteliálně). U některých pacientů mohou být přítomny v RB i nekrózy, srpky a poměrně často i subepiteliální depozita označovaná jako „humps“. Imunofluorescenční kritéria pro C3 dominanci ale nesplňují všechny C3G, zejména v iniciálních fázích onemocnění, a tak se doporučuje (zejména u onemocnění s atypickým průběhem) RB časem opakovat. Načasování RB je velmi důležité, jelikož C3G se často manifestují v průběhu nějaké závažnější infekce. I zde se můžeme s přechodnou dominantní depozicí C3 v RB setkat, ale u parainfekčních typů postižení se depozita časem vymyjí a renální parametry se upravují, zatímco u C3G zůstávají či přibývají. Určitou pomocí může být i vyšetření na přítomnost C4d složky komplementu, jelikož její pozitivita svědčí pro GN mediovanou imunokomplexy.
Klinická manifestace
Atypický hemolyticko‑uremický syndrom
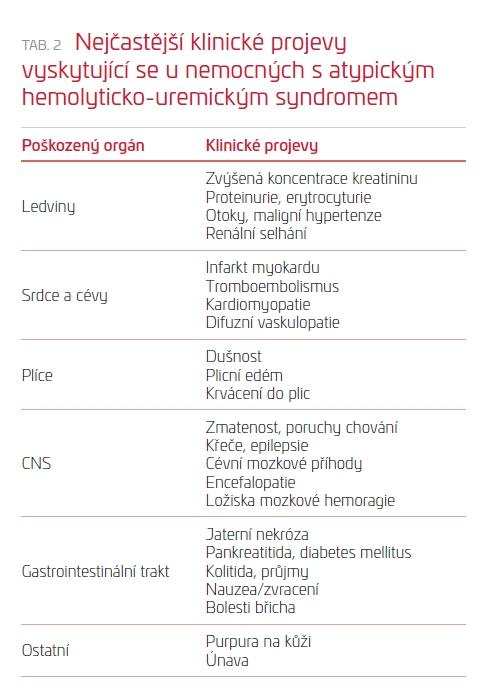
Termín aHUS byl původně používán pro všechny formy hemolyticko‑uremického syndromu (HUS) s výjimkou STEC‑HUS. Současná klasifikace ale reflektuje novější poznatky o etiopatogenezi a mechanismu vzniku těchto onemocnění spolu se znalostí jejich genetického podkladu a spouštěcích faktorů. S ohledem na tyto poznatky někteří klinici používají termín „primární aHUS“ pro stavy spojené striktně s prokázanými odchylkami v alternativní cestě aktivace komplementu (geneticky způsobené, s přítomností protilátek) a na „sekundární aHUS“, tedy ty ostatní. Problém ale je, že jen u zhruba 60 % nemocných se podaří prokázat genetickou abnormalitu přesto, že se jasně chovají jako primární aHUS (relabující či refrakterní formy aHUS). Navíc u řady nemocných s primárním aHUS dochází k manifestaci onemocnění až po zásahu jakéhosi „spouštěče“ a do té doby je choroba asymptomatická. Mezi tyto spouštěče (někdy také nazývané complement amplifying conditions) patří třeba určité typy infekcí, léků, autoimunitních chorob, těhotenství či transplantace (tab. 1). Tyto choroby pak samy mohou být příčinou vzniku časově omezeného sekundárního aHUS. První manifestace TMA u primárních forem aHUS bývá většinou nejzávažnější a může být spojena s vysokou morbiditou (zejména s rizikem renálního selhání u 70 % jedinců), ale i mortalitou. Klinické projevy onemocnění jsou uvedeny v tab. 2. Dosud není zcela jasné, zda tyto klinické projevy jsou způsobeny aktivací komplementu, TMA či jinými faktory, jako je maligní hypertenze či uremie.

C3 glomerulopatie
C3 glomerulopatie je onemocnění s chronickým průběhem, které má ve většině případů progresivní charakter. Typicky se projevuje trvalou aktivitou alternativní cesty komplementu (snížení hodnoty C3 složky v séru) s desetiletým přežíváním pacientů s onemocněním ledvin přibližně v 50 %.3 Nejčastějšími nálezy v moči je hematurie a jiná než nefrotická proteinurie (35 %), nefrotický syndrom (35 %), ale někdy se objevuje i nefritický syndrom a u 20 % jedinců jde o poměrně rychle progredující glomerulonefritidu (GN). Často bývá přítomna i těžká arteriální hypertenze (50–80 %). Jako extrarenální manifestace je u DDD popisován výskyt parciální lipodystrofie a přítomnost drúz v retině, což je oboje důsledkem aktivace komplementu. Získaná parciální lipodystrofie se nejčastěji vyskytuje u jedinců, kteří mají pozitivní C3 nefritický faktor (C3Nef; protilátka proti C3 konvertáze).
Laboratorní testy
Atypický hemolyticko‑uremický syndrom
Po potvrzení přítomnosti TMA je na prvním místě potřeba vyloučit TTP stanovením aktivity ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) a případně přítomnost protilátek proti ADAMTS13; diagnózu TTP potvrzuje jeho koncentrace pod 10 % běžné aktivity. Dále bychom měli vyšetřit expresi CD46 na povrchu makrofágů a odebrat sérum na stanovení přítomnosti protilátek proti komplementárnímu faktoru H (anti‑CFH). Současně vylučujeme další příčiny HUS, zejména STEC‑HUS (kultivace stolice na přítomnost E. coli s produkcí shigatoxinu) a jiné sekundární formy aHUS (viz tab. 1). Po stanovení diagnózy „primární aHUS“ by mělo následovat genetické vyšetření nemocných, jehož znalost není podmínkou zahájení specifické terapie. Nezbytností ale je znalost výsledků genetiky v případě, že onemocnění progredovalo do terminálního selhání ledvin a plánujeme u něj transplantaci ledviny (TxL), zejména pokud by mělo jít o živého dárce. Vyšetřování anti‑CFH protilátek by mělo být prováděno opakovaně (zejména u dětí a v období před TxL).
C3 glomerulopatie
Typickým laboratorním nálezem u nemocných s C3G je snížení plazmatické koncentrace C3 složky komplementu (až u 75 % nemocných). Rovněž u aHUS, STEC‑HUS či jiných chorob ledvin (např. parainfekčních) může být koncentrace C3 v plazmě snížená, takže jde o ukazatel nespecifický, nicméně tento nález diagnózu výrazně podporuje. U nemocných s DDD by měla být vyloučena přítomnost C3Nef. Pro aktivaci alternativní složky komplementu také svědčí snížená aktivita složky CH50 komplementu. U některých pacientů s C3G může být onemocnění spojeno s přítomností anti‑CFH protilátek anebo s pozitivitou monoklonálních lehkých řetězců kappa či lambda v séru. Genetické vyšetření u nemocných s C3G není rutinně doporučováno, nicméně může napomoci v případě rozhodování o léčbě (blokáda komplementu versus imunosupresivní léčba). Jednoznačně namístě je ale pak u onemocnění s rodinným výskytem, jasným genetickým defektem anebo v případě plánování příbuzenské TxL. Riziko rekurence C3G ve štěpu se totiž odhaduje až na 50 %.
Terapie
Atypický hemolyticko‑uremický syndrom
Všichni nemocní s primárním aHUS jsou vhodnými kandidáty pro léčbu inhibitory komplementu (např. ekulizumab; monoklonální protilátka proti C5). Tato léčba je účinná a vede k rychlému vymizení známek TMA a ke vzestupu počtu trombocytů. Dávkování léku vycházelo z několika studií, na jejichž základě se vygenerovalo optimální schéma podávání.4,5 Při dodržování doporučeného dávkování není potřeba kontrolovat účinnost léku pomocí parametrů ukazujících na inhibici komplementu. Jiná situace ale nastává, pokud bychom chtěli léčbu přerušit či ukončit. Zde jasná doporučení neexistují a účinnost léku je nutné kontrolovat například stanovením aktivity alternativní cesty komplementu (AH50), která by při účinné léčbě měla být nižší než 10 % normy. Léčbu se nedoporučuje vysazovat v době interkurentních infekcí (s výjimkou infekcí s enkapsulujícími organismy), jelikož nemocným hrozí relaps onemocnění. Nemocní s tvorbou anti‑CFH protilátek by měli být iniciálně léčeni také ekulizumabem; současně ale u nich zahajujeme i imunosupresivní léčbu (cyklofosfamid nebo rituximab v indukci; mykofenolát mofetil + kortikosteroidy v udržovací léčbě) a v závislosti na vlivu léčby a poklesu titru protilátek léčbu vysazujeme (ekulizumab; následně pak po šesti měsících je možné zvážit i ukončení imunosuprese). Tam, kde není ekulizumab dostupný, se k léčbě používají výměnné plazmaferézy. Ty lze indikovat i u nemocných s anti‑CFH pozitivním aHUS, na úvod léčby u kriticky nemocných pacientů anebo na překlenutí doby, než je ekulizumab dostupný. Délka trvání léčby je v současné době velmi diskutovaným tématem. V rozhodování o ukončení terapie by měl být zohledněn typ mutace a dále výsledná úroveň renální funkce, aby došlo k optimální a maximální reparaci ledvin a minimalizovalo se riziko relapsu (provedené studie demonstrují vzestup glomerulární filtrace ještě po řadu měsíců od zahájení terapie).
Jelikož ekulizumab zvyšuje riziko infekce meningokokovou meningitidou, je nutné nemocné před zahájením léčby očkovat proti meningokokům (včetně typu B) anebo jim podávat profylaktickou antibiotickou terapii (během léčby ekulizumabem a po dobu 2–3 měsíců po jejím skončení).
Transplantace ledviny by neměla být provedena dříve než za šest měsíců po zahájení dialyzačního léčení (z důvodů možné pokračující reparace renální funkce – viz výše) a samozřejmě za předpokladu, že kompletně vymizejí známky TMA. Rozhodnutí o použití profylaktické terapie blokující komplement v návaznosti na TxL je uvedeno v tab. 3. Transplantace jater je možným terapeutickým přístupem u nemocných s těžkým průběhem onemocnění, s omezenou odpovědí na ekulizumab a s mutací ve faktorech syntetizovaných v játrech (CFH, CFI, C3, CB).6

C3 glomerulopatie
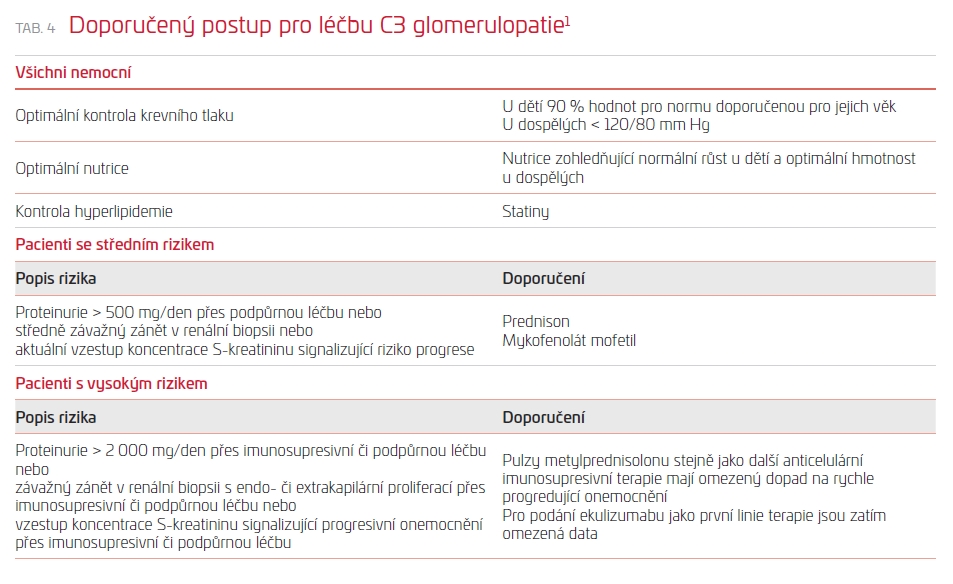
Doporučení pro léčbu C3G jsou bohužel extrémně slabá, a to kvůli naprosté absenci studií. Existuje jen jedna randomizovaná studie, kde se podávaly samotné kortikosteroidy proti placebu u mezangiokapilární GN u dětí, která byla navíc zatížena velkou heterogenitou v terminologii i charakteristice vlastního onemocnění (studie z roku 1992, kdy se o současném dělení a definici C3G moc nevědělo). V jedné retrospektivní studii pak byly demonstrovány účinky mykofenolát mofetilu u selektovaného vzorku jedinců.7 Doporučení pro léčbu C3G jsou uvedena v tab. 4 a představují spíše názor expertů než závěry podložené silnými daty. Existují kazuistická sdělení o účinnosti rituximabu či plazmaterapie u C3G, zejména tam, kde je onemocnění spojeno s přítomností protilátek (C3Nef, anti‑CFH). Probíhají a plánují se studie zaměřující se zejména na blokátory aktivace komplementu.
Literatura
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusion from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int 2017;91:539–551.
- Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2016;31:15–39.
- Servais A, Noël LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulo-pathies. Kidney Int 2012;82:454–464.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic uremic syndrome. N Engl J Med 2013;368:2169–2181.
- Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2 year extensions of phase 2 studies. Kidney Int 2015;87:1061–1073.
- Coppo R, Cavero T, Roman E, et al. Liver transplantation for aHUS: still needed in the eculizumab era? Pediatr Nephrol 2016;31:759–768.
- Rabasco C, Cavero T, Romana E, et al. Effectiveness of mycophenolate mofetil in C3 glomerulonephritis. Kidney Int 2015;88:1153–1160.
- Kategorie: Přehledové články
- Klíčová slova: atypický hemolyticko‑uremický syndrom; C3 nefropatie; ekulizumab; komplement; trombotická mikroangiopatie