Střádavá onemocnění v nefrologii
Souhrn
Nejčastější střádavé onemocnění postihující ledviny představuje Fabryho choroba. Ostatní střádavá onemocnění jsou vzácná, postihují většinou tubulární funkce ledvin. Fabryho choroba je dědičné metabolické onemocnění ze skupiny lysosomálních střádavých onemocnění na podkladě mutace genu pro enzym α‑galaktosidázu A, který je lokalizován na chromosomu X. Projevuje se hromaděním globotriaosylceramidu s následnou hypertrofickou kardiomyopatií, selháním ledvin, postižením periferního i centrálního nervového systému, kůže, očí i dalších orgánů. Diagnózu stanovíme na základě rodinné anamnézy, klinického obrazu, nízké aktivity enzymu α‑galaktosidázy v plazmě či v leukocytech a potvrdíme ji molekulárně‑genetickým vyšetřením. Jako screeningovou metodu u mužů je výhodné v případě podezření na Fabryho chorobu použít dry blood spot test. Diagnóza je však často stanovena pozdě, protože se na ni nemyslí. Postižení ledvin začíná většinou mikroalbuminurií, která progreduje do proteinurie, a postupně klesá renální funkce. K renálnímu selhání dochází většinou ve čtvrtém decenniu. Existuje specifická enzym‑substituční terapie, která spočívá v nitrožilní aplikaci chybějícího enzymu jedenkrát za dva týdny. Klinické studie prokázaly dobrý účinek, bylo‑li s léčbou započato před rozvinutím ireverzibilního postižení.
Nejčastěji se vyskytujícím onemocněním ze střádání, které nejčastěji postihuje ledviny, je Fabryho choroba. K selhání ledvin dochází většinou až v dospělém věku.Ostatní střádavé choroby postihující ledviny jsou mnohem vzácnější a nejčastěji se projevují sekundárním Fanconiho syndromem, protože jako první jsou postiženy tubulární buňky. Projevy se objevují většinou již v dětském věku (tab. 1).
Fabryho choroba
Fabryho choroba je vzácné dědičné onemocnění patřící mezi lysosomální choroby ze střádání. Je způsobeno mutací genu pro enzym α‑galaktosidázu A, který se účastní odbourávání glykosfingolipidů. Gen je lokalizován na chromosomu X (GLA gen), jedná se tedy o gonosomálně recesivní dědičnost. Mutace způsobuje absolutní nebo relativní nedostatek enzymu a jeho důsledkem je hromadění globotriaosylceramidu (Gb3) v lysosomech buněk různých tkání a jejich poškození.
Prevalence choroby je odhadována na jeden případ na 40 tisíc nově narozených chlapců a na 20 tisíc nově narozených dívek. S chorobou se setkáváme ve všech zemích a postihuje všechny lidské rasy.
Ke střádání dochází již intrauterinně. Akumulace Gb3 vede postupně k poškození tkání a následně k orgánovému selhání. Klasická forma choroby se projevuje již v dětství postižením periferní nervové soustavy (bolestmi, pálením dlaní a chodidel, projevy dráždivého tračníku). Časté je snížené pocení (hypohidróza), u dětí se mohou objevit febrilie nejasné etiologie (občas i hypertermické krize). Později vznikají až u 80 % dospělých kožní léze, tzv. angiokeratomy. Ve druhém a třetím decenniu se přidává postižení srdce (hypertrofická kardiomyopatie, poruchy srdečního rytmu, vzácně i postižení srdečních chlopní) a také postižení ledvin, které vede k jejich terminálnímu selhání. Častěji se vyskytují také cévní mozkové příhody. Nejčastěji jsou popsány ischemické mozkové příhody, a to již ve věku okolo třetího decennia. Může se vyskytovat i mozkové krvácení. Časté jsou projevy vertebrobazilární insuficience. Typické je postižení očí (tzv. cornea verticillata). Na spojivce i sítnici bývají vinuté a dilatované cévy. Postižení zraku u Fabryho choroby zpravidla nevede k významnějšímu poškození vizu, nicméně oční nález je důležitým diagnostickým vodítkem.
Existuje několik set popsaných mutací GLA genu. Většina mutací je jedinečných, až 70 % mutací tvoří záměny bází typu nonsense nebo missense mutací. Až 80 % žen přenašeček má určité příznaky choroby a až u 20 % z nich se mohou vyskytnout život ohrožující symptomy. Proto je v poslední době zvažováno klasifikovat toto onemocnění jako X‑dominantně vázané. U obou pohlaví se mohou vyskytovat také formy, které postihují pouze jeden orgán (např. kardiální nebo renální forma choroby). U takto postižených pacientů bývá přítomna reziduální aktivita α‑galaktosidázy A. Postižení může být velmi variabilní, a to dokonce i v rámci jedné rodiny. Bez léčby má postižení mužů zpravidla progresivní charakter a vede k závažnému orgánovému postižení, k výraznému snížení kvality života a k předčasnému úmrtí. U heterozygotních žen očekávajících mužského potomka je možné provést enzymaticky prenatální diagnostiku z choriových klků již v 10. týdnu gravidity.
Postižení ledvin
Postižení ledvin se vyskytuje u většiny mužů a u části žen. Mikroalbuminurie bývá přítomna v případě těžších forem již u dětí. Svým vývojem může připomínat diabetickou nefropatii.
Depozita Gb3 se ukládají v různých buňkách ledvin (nejdříve v podocytech, dále v epiteliálních buňkách, v tubulárních buňkách hlavně distálního tubulu, v endoteliálních buňkách a v hladkých svalových buňkách arteriol) od raného dětství. Následně dochází k proliferaci mesangiálních buněk, k proliferaci hladkých svalových buněk s vaskulopatií, k ischémii glomerulů s následnou sklerotizací glomerulů a s atrofií tubulů. Charakteristickým nálezem je vakuolizace v podocytech a v epiteliálních buňkách při vyšetření vzorku renální biopsie ve světelné mikroskopii (následek fixace). V elektronovém mikroskopu jsou vakuoly vyplněny depozity Gb3.
Klinicky se nejprve objeví mikroalbuminurie, která progreduje do proteinurie, ale málokdy se setkáváme s nefrotickou proteinurií. Mikroskopická hematurie je vzácná. Depozita Gb3 byla zjištěna již u malých dětí bez mikroalbuminurie. Mikroalbuminurie a renální funkce by se měly u pacientů stanovovat od období adolescence pravidelně jedenkrát ročně. Proteinurie koreluje s renální funkcí a přispívá k jejímu poklesu. Onemocnění má progresivní charakter a dospěje k terminálnímu selhání ledvin. K renálnímu selhání dochází mezi třetím až pátým decenniem (v průměru 36.–38. rok života). Selhání ledvin bylo ale popsáno i u 16letého pacienta. Až u 50 % pacientů se vyskytují také kortikální a parapelvické cysty. Arteriální hypertenze se rozvíjí u více než 60 % pacientů, bývá spíše mírnější a většinou je přítomna až od stadia 3 chronické renální insuficience. Nutné je ale dávat pozor i na hypotenzi kvůli autonomní dysfunkci, která je spojena s vyšším kardiovaskulárním a renálním rizikem. Postižení tubulů v počáteční fázi bývá spojeno se ztrátou koncentrační a diluční schopnosti ledvin (isostenurie, hypostenurie), i při normální funkci ledvin může dojít k rozvoji metabolické acidózy. Výjimečně se při akumulaci Gb3 v buňkách proximálního tubulu rozvine Fanconiho syndrom.
Renální biopsie má význam u pacientů s již diagnostikovanou Fabryho chorobou ke stanovení stupně glomerulosklerózy a intersticiální fibrózy, které dlouhodobě korelují s poklesem renální funkce. Dále se zvažuje při rychlém poklesu renální funkce, při suspekci na jinou glomerulopatii (př. diabetickou nefropatii, IgA nefropatii). Je zvažována u mladých pacientů s mikroalbuminurií před zahájením enzymatické terapie k posouzení depozit Gb3 nebo následně při hodnocení účinku enzymatické léčby v rámci studií.
Pokles renální funkce závisí na pohlaví (rychlejší u mužů), na proteinurii, na hypertenzi a progreduje rychle od stadia 3 chronické renální insuficience. Při léčbě dialýzou přežívá v Evropě tři roky 60 % všech pacientů s Fabryho chorobou (diabetici – 53 %, nediabetici – 74%). Pacienti umírají především na kardiovaskulární komplikace. Přežití po transplantaci štěpu je velmi dobré (5 let přežívá 81 % nemocných). Mortalita prudce roste po deseti letech po transplantaci. I prvních pět let po transplantaci pacientům hrozí vyšší riziko náhlého úmrtí.
Diagnostika
Často je velká prodleva mezi prvním příznaky a diagnózou onemocnění. U mužů byly zjištěny první příznaky v devíti letech a diagnóza byla stanovena v průměru až ve 23 letech, u žen byly přítomny první příznaky ve věku 13 let a diagnóza stanovena v průměru až ve 32 letech (z Registru Fabryho choroby). Důležitá je echokardiografie, popř. magnetická rezonance srdce, která pomůže posoudit stupeň fibrózy myokardu (typicky se fibróza objevuje v posterolaterální oblasti levé komory). V nefrologii nejsou nálezy k diagnostice Fabryho choroby specifické, někdy je diagnóza stanovena náhodně z renální biopsie ( jednoznačný nález inkluzí v elektronové mikroskopii). Diagnózu potvrdíme stanovením aktivity enzymu α‑galaktosidázy A v plazmě nebo v leukocytech. U žen se však hodnoty aktivity enzymu mohou překrývat s hodnotami u všeobecné populace a diagnózu je třeba potvrdit molekulárně‑genetickým vyšetřením.
Screening
U mužů je možné využít screeningového vyšetření metodou suché kapky (dry blood spot test, DBS), které je pozitivní při aktivitě enzymu nižší než 35 % normy. Stanovení aktivity enzymu i molekulárně‑genetické vyšetření provádí laboratoře Ústavu dědičných metabolických poruch při 1. LF UK a VFN v Praze. Toto vyšetření je velmi rychlé a levné a velmi přínosné. Test provedený metodou suché kapky je schopen zjistit diagnózu jen u jedné třetiny heterozygotních žen, proto se dnes ke screeningu heterozygotek nepoužívá. U suspektních žen je nutné rovnou provádět mutační analýzu; DBS test je výhodné využít u rizikových skupin pacientů. V roce 2003 byl pomocí DBS testu proveden screening mezi hemodialyzovanými pacienty i v České republice.1 Zjištěná prevalence Fabryho choroby u hemodialyzovaných pacientů v ČR byla tehdy 0,26 % u mužů a 0,05 % u žen. U tří z pěti pacientů nebyla původně provedena renální biopsie (dva pacienti vedeni jako diabetická nefropatie, jeden jako tubulointersticiální nefritida), u dalšího pacienta byla pomocí imunofluorescence (IF) stanovena IgA nefropatie a nebyla provedena elektronová mikroskopie. U jediné pacientky s nalezenou mutací byla původně v renální biopsii diagnostikována membranoproliferativní glomerulonefritida. Podobná prevalence (okolo 0,2 %) byla zjištěna i v jiných evropských zemích.
I nyní se screening provádí na některých dialyzačních pracovištích u pacientů s nejasnou diagnózou renálního selhání. Dále se screening provádí i u pacientů s nejasnou renální diagnózou v nefrologických ambulancích. Test pomocí DBS by měli podstoupit muži mladší 50 let bez spolehlivé renální diagnózy s chronickou renální insuficiencí stupně 2 a více nebo s proteinurií (ev. s mikroalbuminurií). Screening u žen prováděný pomocí mutační analýzy GLA genu je doporučen bez ohledu na věk u pacientek s nejasným renálním onemocněním a s příznaky Fabryho choroby.2
Jsou pacienti, u kterých nemusejí být jasně vyjádřeny systémové příznaky a může převládat renální nebo kardiální postižení. Postižení periferního nervového systému může imitovat diabetickou polyneuropatii. Pacienti s diabetem mohou současně trpět Fabryho chorobou. U našich pacientů se vyskytla i koincidence s IgA nefropatií. Při diagnostice z renální biopsie je vždy nutné mít vzorek z biopsie vyšetřen i v elektronovém mikroskopu.
Léčba
Substituční enzymová terapie (enzyme replacement therapy, ERT) je dostupná teprve v posledním desetiletí. Na trhu jsou k dispozici dva přípravky – β‑galaktosidáza (Genzyme), která se podává v dávce 1 mg/kg, a α‑galaktosidáza (Shire), která se podává v dávce 0,2 mg/kg. Rozdíly v jejich složení jsou minimální, liší se pouze v glykosylaci. Lék se podává v nitrožilní infuzi 1krát za 14 dní. Lék dokáže vyčistit tkáně od Gb3 a zpomalit progresi onemocnění. Dochází ke zmírnění neuropatických bolestí, ke zpomalení poklesu glomerulární filtrace, ke zpomalení progrese hypertrofie myokardu. Klinické studie prokazují i zlepšení kvality života a účinek na prognózu nemocných (pokles výskytu závažných kardiovaskulárních a renálních příhod). Při šestiměsíčním podávání placeba pacientům s Fabryho chorobou představoval pokles odhadované glomerulární filtrace (estimated glomerular filtration rate, eGFR) –7 ml/min/1,73 m2. Při léčbě α‑galaktosidázou po dobu 1–4,5 roku dosáhl pokles poloviční hodnoty, a to –2,9 ml/min/1,73/m2. Nejdéle trvající – desetiletá – studie s β‑galaktosidázou prokázala významné zpomalení poklesu eGFR hlavně u pacientů mladších 25 let s lehkým renálním postižením (proteinurie < 0,5 g/24 h, < 50 % sklerotických glomerulů), u kterých představoval pokles eGFR při podávání ERT za rok –1,89 ml/min/1,73 m2.3 Ukončení ERT je zvažováno u pacientů se selháním ledvin s kontraindikovanou transplantací a s pokročilým srdečním selháním (NYHA IV), pokud je pravděpodobná doba dožití kratší než jeden rok.
Mezi nejčastější nežádoucí účinky patří infuzní reakce, které vznikají zpravidla na alergickém podkladě. Vyskytují se ve více než v 5 % případů. Nejobvykleji se jedná o třesavky, zimnice, febrilie, bolesti hlavy či o bolesti kloubů a nevolnost. Tyto reakce zpravidla odezní po zpomalení infuze a podání antipyretik, antihistaminik či kortikoidů. Někdy se vyskytne kopřivka, svědění kůže, dušnost nebo otok hrtanu. Nejzávažnější reakcí je anafylaxe.
Z renálního hlediska je určitě důležité myslet na ovlivnění proteinurie a progrese chronické renální insuficience inhibitory ACE (angiotensin konvertujícího enzymu) a sartany. K dispozici však není žádná kontrolovaná klinická studie. Tahir a kol. poukázali na fakt, že ERT samotná nedokázala kontrolovat proteinurii a že teprve po přidání inhibitorů ACE nebo sartanů do léčby došlo k její regresi.4 Všichni pacienti trpící Fabryho chorobou by měli být při proteinurii (nejlépe již při zachycení mikroalbuminurie) léčeni inhibitory ACE nebo sartany. Nižší proteinurie je spojena i s lepším ovlivněním renální prognózy finančně náročnou ERT.
Péče o pacienty s Fabryho chorobou je soustředěna do Centra pro Fabryho chorobu na II. interní klinice kardiologie a angiologie VFN a 1. LF UK v Praze. Léčbu je možné zahájit jen se souhlasem příslušné pojišťovny a vedení nemocnice po posouzení indikace k léčbě. Léčba se zahajuje po zjištění symptomů nebo jasných laboratorních známek onemocnění. U pacientů mužů s klasickou formou Fabryho choroby je možno od věku 16 let zahájit ERT, ačkoli nejsou přítomny známky orgánového poškození a pacienti jsou bez symptomů. Infuze s enzymem jsou podávány v nejbližší nemocnici, nejčastěji v ambulantním režimu v dialyzačním středisku. Nyní probíhá několik studií s chaperony, které vazbou na α‑galaktosidázu zvyšují její účinnost. Fungují jen u pacientů s některými missense mutacemi. Výhodou je možnost podávání v tabletách. Výsledek šestiměsíční studie s chaperonem Migalastat nebyl zcela jednoznačný.5
Velkou naději pro pacienty s Fabryho chorobou představuje genová terapie. Problémem však zůstává imunitní reakce, která vede k poškození vektoru.
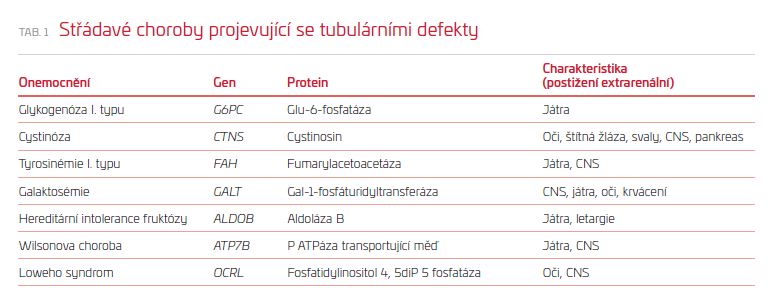
Střádavé choroby projevující se renálními tubulárními defekty
Cystinóza
Jedná se o nejčastější vrozenou příčinu Fanconiho syndromu. Cystinóza je choroba, která vzniká v důsledku intralysosomální akumulace aminokyseliny cystinu, k níž dochází následkem defektního transportu cystinu přes lysosomální membránu. Podle klinického průběhu se rozlišují tři formy cystinózy – nejtěžší infantilní cystinóza, juvenilní cystinóza a okulární cystinóza. Onemocnění je přenášeno autosomálně‑recesivní dědičností (AR). Prevalence infantilní formy je asi 1 : 200 000. Gen pro cystinosin (tzv. CTNS gen) je umístěn na 17. chromosomu. Bylo popsáno více než 100 různých mutací. Nejčastější mutací vyskytující se u evropských pacientů je rozsáhlá delece (od exonu 1 do exonu 10), která byla zjištěna u 76 % evropských pacientů. Pacienti s juvenilní nebo s okulární cystinózou mají alespoň jednu missense mutaci ve funkčně nedůležité oblasti proteinu. Cystinosin je lysosomální transportér, který je vysoce specifický pro L‑cystin a je řízen protony. Prvními klinickými projevy klasické infantilní nefropatické cystinózy jsou projevy Fanconiho syndromu, který se většinou plně rozvine do jednoho roku věku dítěte. Rachitida a porucha růstu jsou velmi běžné, později se vyvíjí porucha renálních funkcí se vznikem chronického selhání ledvin. Do věku tří let se rovněž vyvíjejí fotofobie, vzácně i poruchy zraku. Mezi pozdní komplikace patří hypofunkce štítné žlázy, diabetes mellitus (většinou až po transplantaci ledviny), hepatosplenomegalie, dysfagie, respirační insuficience a ulcerace rohovky.
Diagnózu lze stanovit na podkladě biochemického průkazu zvýšených nitrobuněčných koncentrací cystinu v leukocytech. Oční projevy – přítomnost krystalků cystinu v rohovce – lze prokázat vyšetřením štěrbinovou lampou. Dále je možno provést mutační analýzu genu CTNS. Z technických důvodů zatím není dostupný neonatální screening. V současné době tvoří základ terapie podávání cysteaminu (Cystagon – nutno podávat jednou za 6 hodin, Procysbi – podává se jednou za 12 hodin). Do očí se podávají kapky s cysteaminem. S léčbou je nutné začít co nejdříve. Terapie cysteaminem je často doprovázena zažívacími obtížemi ze zvýšené sekrece gastrinu a bohužel nevede k úpravě Fanconiho syndromu. Oddálí dobu selhání ledvin o 6–10 let. Po transplantaci je renální prognóza velmi dobrá, je ale nutno pokračovat v léčbě cysteaminem z důvodů extrarenálních projevů.
Glykogenóza typu I (nemoc z ukládání glykogenu) – von Gierkeho choroba
Nemoc z ukládání glykogenu (glycogen storage disease, GSD) zahrnuje 12 různých genetických defektů zasahujících do metabolismu glykogenu s primárním postižením jater, svalů či obou struktur. Dochází k epizodám těžkých hypoglykémií, protože je poškozena jak glykogenolýza, tak glukoneogeneze. Glykogenóza typu I je jedinou GSD, při které jsou primárně postiženy i ledviny. Jde o vzácné AR onemocnění (výskyt 1 : 100 000). Biochemickou podstatou poruchy u GSD‑Ia je deficit aktivity glukózo‑6‑fosfatázy v játrech, v ledvinách a ve střevech, s průvodním ukládáním glykogenu v těchto orgánech.
Onemocnění lze stanovit sníženou aktivitou enzymu v jaterní biopsii. Renální postižení u GSD‑I zahrnuje sekundární Fanconiho syndrom, renální tubulární acidózu distálního typu, hyperurikemii, případně fokálně segmentální glomerulosklerózu. Častěji je popisována i hyperkalciurie s nefrokalcinózou, která může vést k progresi renální insuficience. U části pacientů se rozvíjejí cysty ledvin a dochází k selhání ledvin. Terapie spočívá v podávání syrového kukuřičného škrobu a maltodextrinů. Nutná jsou častá jídla během dne a na noc se začíná podávat postupně se uvolňující kukuřičný škrob, který umožní pacientům spát až 7 hodin v noci bez jídla. Ve stravě je nutné eliminovat galaktózu a fruktózu, omezuje se příjem tuků.
Galaktosémie
Galaktosémie je AR onemocnění metabolismu galaktózy. Onemocnění je nejčastěji způsobeno sníženou aktivitou enzymu galaktózo‑1‑fosfáturidyltransferázy (incidence 1 : 62 000 živě narozených dětí), jehož gen je lokalizován na 9. chromosomu. U postižených dětí, které konzumují mléko (nejběžnější zdroj galaktózy v dietě), se vyvíjejí trávicí obtíže (zvracení, průjem), děti neprospívají. Mohou být přítomny známky hemolýzy, žloutenka z přítomnosti nekonjugovaného bilirubinu, vyvíjí se splenomegalie a katarakta. Galaktosémie způsobuje zvýšené vylučování aminokyselin a albuminu močí, z hlediska ztrát cukrů do moči je prokazatelná galaktosurie. Potvrzení suspektní diagnózy lze uskutečnit průkazem deficitu enzymu. Léčba spočívá v eliminaci galaktózy z diety.
Hereditární intolerance fruktózy
Hereditární intolerance fruktózy (cukrovinky) je další vrozená AR forma poruchy metabolismu sacharidů, která je spojena s obrazem Fanconiho syndromu.
Tyrosinémie
Hereditární tyrosinémie typu I představuje defekt metabolismu tyrosinu postihující játra, ledviny a periferní nervy. Podstatou onemocnění je deficit fumarylacetoacetátové hydrolázy (FAH), který se dědí AR typem dědičnosti. Gen pro tento enzym je lokalizován na 15. chromosomu. Snížená nebo chybějící aktivita FAH je spojena s hromaděním maleylacetoacetátu a fumarylacetoacetátu v postižených tkáních. Tyto sloučeniny mohou působit jako alkylační látky. Hlavním poškozeným orgánem bývají játra, porucha funkce proximálního tubulu je prokazatelná u všech pacientů s tyrosinémií. Zavedení dietního režimu o nízkém obsahu fenylalaninu a tyrosinu zmírňuje poruchu tubulární funkce. Dále se používá nitisinon, který inhibuje tvorbu maleylacetoacetátu a fumarylacetoacetátu. U pacientů se selháním jater se provádí transplantace jater, která vede i ke korekci Fanconiho syndromu.
Wilsonova choroba
Wilsonova choroba je vrozená porucha metabolismu mědi, která vede k multiorgánovému postižení (postižení jater, extrapyramidové příznaky, psychiatrické poruchy). Wilsonova choroba představuje defekt ATPázy typu P, transportéru pro měď v játrech. Tento defekt vede k poruše vylučování mědi a k zabudování mědi do ceruloplasminu. Tyto abnormality způsobují nadměrné hromadění mědi v játrech s následným ukládáním i do mozku, dále do rohovky a renálního proximálního tubulu. Nadměrné hromadění mědi v ledvinách vede k tubulární dysfunkci a u některých pacientů k vývoji úplného Fanconiho syndromu. Sérové koncentrace ceruloplasminu jsou sníženy u 90 % pacientů s Wilsonovou chorobou. Výrazně zvýšené hodnoty mědi v moči rovněž přispívají ke stanovení diagnózy, zvláště stoupají‑li při podání D‑penicilaminu. Koncentrace mědi v játrech (diagnostika z jaterní biopsie) jsou zvýšené u neléčených pacientů. Je možné provést i molekulárně‑genetickou diagnostiku onemocnění. Terapie D‑penicilaminem může vést pozvolna k úpravě ledvinných poruch i ovlivnit průběh jaterního a neurologického postižení. Většina i léčených pacientů s Wilsonovou chorobou zůstává sledována nefrology pro často přetrvávající, i když mírné projevy renální tubulární acidózy.
Loweho syndrom
Pro Loweho syndrom (okulocerebrorenální syndrom) je typická konstelace vrozené oboustranné katarakty a glaukomu, mentální retardace, hypotonie a renální abnormality. Gen pro Loweho syndrom, lokalizovaný v oblasti Xp24‑26 (gen OCRL1), je přenášen X‑recesivním typem přenosu. Onemocnění se však výjimečně projevuje i u žen. Tento gen kóduje fosfatidylinositol‑4,5‑bifosfát‑5‑fosfatázu lokalizovanou v Golgiho komplexu. Zpočátku se renální postižení projevuje změnami v rámci Fanconiho syndromu, proteinurie se objevuje v prvních měsících po narození. Často se vyskytuje hyperkalciurie, nefrolitiáza, nefrokalcinóza. Později dochází k poklesu glomerulární filtrace se vznikem chronického selhání ledvin (ve 3.–4. dekádě). Léčba je pouze symptomatická.
Vyskytují se i formy bez postižení mozku a oka, které jsou označovány jako Dentova choroba typu 2. Mutace jsou opět v genu OCRL1, ale pouze v prvních sedmi exonech.
Deficit lecithin‑cholesterol acyltransferázy
Deficit lecithin‑cholesterol acyltransferázy (LCAT) je vzácné AR onemocnění, které se projevuje korneálními opacitami, anémií, dyslipidémií (koncentrace HDL cholesterolu jsou velmi nízké nebo neměřitelné) a v časné dospělosti dochází k rozvoji proteinurie a renální insuficience s renálním selháním, které nastává ve věku mezi 3.–4. decenniem. Při deficitu LCAT je přítomna porucha esterifikace cholesterolu a dochází k ukládání lipidů v různých orgánech. Renální postižení může být potvrzeno z renální biopsie, ve světelné mikroskopii je přítomna expanze mesangia a ztluštění glomerulární bazální membrány (GBM), po speciálním nabarvení jsou patrné nepravidelné vakuoly. Výsledek vyšetření pomocí imunofluorescence je negativní, v elektronovém mikroskopu jsou patrná elektron‑denzní depozita v mesangiu a v glomerulární bazální membráně.
Nyní je dostupná pouze symptomatická léčba hypertenze a léčba statiny. Probíhají studie s ERT.6
Literatura
- Merta M, Reiterová J, Ledvinová J, et al. A nationwide blood spot screening study for Fabry disease in the Czech Republic haemodialysis patient population. Nephrol Dial Transplant 2007;22:179–186.
- Terryn W, Cochat P, Froissart R, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant 2013;28:505–517.
- Germain DP, Charrow J, Desnick RJ, et al. Ten‑year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52:353–358.
- Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and Fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase‑beta. J Am Soc Nephrol 2007;18:2609–2617.
- Germain DP, Hughes DA, Nicholls K. Treatment of Fabry´s disease with the pharmacologic chaperone Migalastat. N Engl J Med 2016;375:545–555.
- Shamburek RD, Bakker‑Arkema R, Auerbach BJ. Familial lecithin: cholesterol acyltransferase deficiency: First‑in‑human treatment with enzyme replacement. J Clin Lipidol 2016;10:356–367.
- Kategorie: Přehledové články
- Klíčová slova: enzymatická substituční léčba; Fabryho nemoc; Fanconiho syndrom; lysosomální střádavá onemocnění; renální selhání; α‑galaktosidáza A